Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Masaru Tanaka and Version 4 by Masaru Tanaka.
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the most common neurodegenerative diseases (NDs), presenting a broad range of symptoms from motor dysfunctions to psychobehavioral manifestations. A common clinical course is the proteinopathy-induced neural dysfunction leading to anatomically corresponding neuropathies. However, current diagnostic criteria based on pathology and symptomatology are of little value for the sake of disease prevention and drug development.
- neurodegenerative diseases
- Alzheimer’s disease
- Parkinson’s disease
- multiple sclerosis
- Huntington's disease
- neuroinflammation
- amyloid
- kynurenine
- stroke
- antioxidant
1. Introduction
Neurodegenerative diseases (NDs) encompass a range of progressive and often incurable conditions which affect the central nervous system (CNS), leading to neurodegeneration and eventually to neural cell death, which cause a broad spectrum of symptoms from motor dysfunctions to psychobehavioral manifestations such as ataxia and dementia [1]. Alzheimer’s disease (AD) and Parkinson disease’s (PD) are the most common NDs. AD represents approximately 60%–70% of about 50 million people worldwide suffering from dementia, which is a major cause of disability and dependency among the elderly. More than 10 million people worldwide suffer from PD, and the incidence increases with age [2]. Other NDs include multiple sclerosis (MS), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), Creutzfeldt–Jakob disease (CJD), human immunodeficiency virus (HIV)-associated neurocognitive disorders (HAND), and stroke-induced secondary neurodegeneration (SND), among others. [3]. Neurological diseases are the leading causes of disability-adjusted life years and second leading cause of deaths after cardiovascular diseases globally [2]. The names of the diseases labeled in honor of distinguished physicians, subtyping according to pathological and clinical findings, overlapping histopathological classification, and numerous molecular discoveries all obscure a fundamental perspective of the diseases and may hamper finely tuned research into pathogenesis and new drug discovery of NDs [4][5][6][7][8][4,5,6,7,8].
Neurodegeneration, a structural and/or functional loss of neurons in the CNS, is a common pathognomonic finding of the brain regions reflecting impairment and dysfunction of the corresponding motor, autonomic, and/or cognitive nervous systems [9]. The neurodegenerative lesions of postmortem brain specimens correlate well with structural and functional imaging studies. The regional patterns of the brain shrinkage may help identify affected domains and diagnose diseases by magnetic resonance imaging (MRI) and positron emission tomography (PET) [10].
Abnormal accumulation of proteinaceous materials in and around neurons is a histopathological hallmark in the degenerative neural tissues. Amyloid-β (Aβ) protein, tau protein, alpha-synuclein (α-syn), transactive response DNA-binding protein (TDP)-43, and myelin debris are common deposits [7]. Most NDs present the inflammatory reactions by activation of the innate immune response factors, as well as the adaptive immune response factors. Neuroinflammation is at least one of the final common pathways leading to neurodegeneration. In degenerative brain tissues, activated glial cells and astrocytes cause accumulation of the abnormal proteins, potentiating further neural injuries [11].
2. The Etiological Links behind Neurodegenerative Diseases: Tryptophan and Bioactive Kynurenines
Disturbance of tryptophan metabolism was reported in NDs [12][93]. Tryptophan is an essential amino acid which is used for protein synthesis and is a precursor to biosynthetic compounds, such as serotonin, melatonin, and nicotinamide adenine dinucleotide (NAD+), among others. A majority of research focused on areas of the serotonin pathway, but increasing attention is paid to the KYN pathway, which produces various bioactive compounds associated with inflammation, the immune system, the nervous system, and psychiatric diseases [13][94]. More than 95% of tryptophan is metabolized in the KYN pathway except for protein synthesis. Tryptophan (TRP) is converted to KYN by the hepatic rate-limiting tryptophan 2,3-dioxygenase (TDO) and ubiquitous rate-limiting indoleamine 2, 3-oxygenase (IDO) 1, which are induced by cortisol, and IFN-α, IFN-γ, and TNF-α, respectively. KYN is converted to anthranilic acid (AA) by the kynureninase, 3-hydroxy-l-kynurenine (3-HK) by the KYN-3-monooxygenase (KMO), and kynurenic acid (KYNA) by KYN aminotransferases (KATs). KATs also convert 3-HK to xanthurenic acid (XA). KYNA is an antagonist at the NMDA receptor. XA is converted to cinnabarinic acid (CA) by autoxidation. AA and 3-HK are converted to 3-hydroxyanthranillic acid (3-HAA) and further to picolinic acid (PA) and quinolinic acid (QA). 3-HK and QA are agonists at the NMDA receptor. QA is converted to NAD+, which is a feedback inhibitor of TDO [14][95] (Figure 12).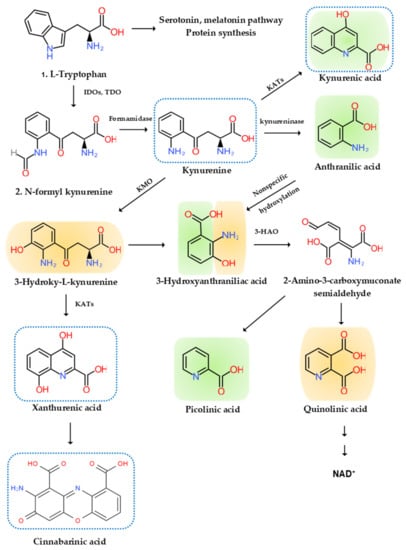
Figure 12. l-Tryptophan metabolism and the kynurenine pathway. The indole ring of l-tryptophan (TRP) is oxidized by the TRP dioxygenase (TDO) and the indolamine-2,3-dioxygenase (IDO) to produce N-formyl kynurenine (KYN). N-formyl KYN is converted by the formamidase to KYN, a substrate of three downstream metabolites: anthranilic acid (AA) by the kynureninase, 3-hydroxy-KYN (3-HK) by the KYN-3-monooxygenase (KMO), and kynurenic acid (KYNA) by KYN aminotransferases (KATs), which also convert 3-HK to xanthurenic acid (XA). XA is converted to cinnabarinic acid (CA) by autoxidation. AA and 3-HK are converted by 3-hydroxyanthranilate oxidase to 3-hydroxy-AA (3-HAA). 3-HAA is converted by 3-hydroxyanthranilate dioxygenase to 2-amino-3-carboxymuconate semialdehyde, which is further transformed into picolinic acid (PA) and quinolinic acid (QA). QA is further converted to nicotinic acid, nicotinic acid adenine dinucleotide, and nicotinamide dinucleotide (NAD+). Neurotoxic, oxidative KYNs are shown in orange color, neuromodulartory anti-inflammatory and antioxidant KYNs are shown in green color, and aryl hydrocarbon receptor agonists are represented by a blue dotted line.
2.1. Tryptophan
Aberrant levels of TRP and disturbance of TRP metabolism are observed in patients suffering from NDs. Low circulating TRP levels are associated in elderly patients with NDs such as AD, PD, and HD [15][96]. TRP supplement was reported to enhance cognitive functions. Short-term TRP supplements improved serial attentions and reaction times and abstract visual memory, while chronic supplementation increased facial recognition memory and decreased baseline startle responsivity [16][97]. A TRP-fortified diet enhanced memory processes without improving the mood states in patients with MS [17][98].2.2. Bioactive Kynurenines
The KYN pathway produces several small receptor agonists and bioactive molecules with a broad range of activities including neurotoxic, neuroprotective, anti-inflammatory, oxidative, antioxidative, and immune properties.2.2.1. Neurotoxic Kynurenines
QA is an excitotoxic NMDA receptor agonist which elicits hippocampal lesions like that of HD. Injections of QA into the striatum of rats induce selective degeneration of intrinsic striatal neurons with striatal atrophy and the lateral ventricle expansion, sparing intrinsic glial cells and myelinated axons of the internal capsule [13][94].2.2.2. Neuromodulatory Kynurenines
KYNA is a receptor antagonist of ionotropic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainite, and NMDA receptors [18][99]. Depending on the dose, however, KYNA exerts different actions at AMPA receptors. The micromolar concentrations are inhibitory, while the nanomolar concentrations are facilitatory by allosteric modulation of the AMPA receptor [19][20][100,101]. The actions of KYNA at the α-7 nicotinic acetylcholine receptor are controversial [21][102]. KYNA reduces glutamate release by binding to the G-protein-coupled receptor 35 (GPR35) in the microglial cells, macrophages, and monocytes to [13][94]. PA was shown to be neuroprotective. PA protects against QA- and kainic acid-induced neurotoxicity in the brain [22][103]. However, PA blocks the neurotoxic effects, but not the excitatory effects of QA. The mechanism of its anti-neurotoxic action is unclear but may be involved in zinc chelation or inhibition of nitric oxide synthase [23][104].2.2.3. Anti-Inflammatory Kynurenines
KYNA binds to the GPR35 expressed in the glia, macrophages, and monocytes to reduce pro-inflammatory cytokine release in cell lines [24][105]. AA and its 5-hydroxylated metabolites may possess potential anti-inflammatory properties. AA is metabolized to 3-HAA by a microsomal hydroxylase in mammalian liver. The anti-inflammatory reaction of AA and its related metabolites are associated with the fact that AA is a precursor of some nonsteroidal anti-inflammatory drugs such as mefenamic acid and diclofenac [25][106]. AA and 3-HAA were found to suppress pro-inflammatory cytokine IFN-γ, T- and B-lymphocyte cell proliferation, Th1 cell activity, and neurotoxicity induced by IL-1 or IFN-γ. They also invoke anti-inflammatory cytokine IL-10 [26][107]. PA can influence the immune response and has antifungal, antitumoral, and antibacterial activities [27][108].2.2.4. Reactive Oxygen Species
3-HK, 3-HAA, and QA are neurotoxic. 3-HK and 3-HAA generate ROS and might play a role in the regulation of OS [28][109]. An elevation of 3-HK levels was related to excitotoxic injury and is observed in patients with NDs [29][110]. The neurotoxic effect of the intermediates 3-HK and 3-HAA involves the generation of superoxide anion and hydrogen peroxide, which contribute to the oxidative processes implicated in the pathophysiology of meningitis [30][111]. 3-HK is the only KYN metabolite that is increased in plasma from vitamin B6-deficient subjects, suggesting that pyridoxal phosphate (PLP)-dependent enzymes are involved in its clearance [31][112]. The production of 3-HK and other TRP metabolites which filter ultraviolet light contribute to a gradual increase in yellowish pigmentation in the lens with age [32][113]. QA is also a free-radical metabolite. The pro-inflammatory cytokine IFN-γ activates IDO, formamidase, and KMO activities in human microglial cells and macrophages, resulting in increased QA synthesis. QA concentrations in the brain were reduced by anti-inflammatory steroid agent dexamethasone after immune stimulation [33][114].2.2.5. Antioxidant
KYNA is an antioxidant metabolite that scavenges ROS observed in various in vitro models and suppresses overshooting inflammation preventing tissue damage. KYNA can reduce FeSO4-triggered ROS toxicity primarily involving superoxide and hydrogen peroxide production [28][34][109,115]. Insufficient KYNA production may contribute to tissue damage and cell proliferation in inflammatory response in NDs.2.2.6. Immune Kynurenines
KYN, KYNA, xanthurenic acid, and cinnabarinic acid (CA) bind to the cytosolic aryl hydrocarbon receptor (AhR) transcription factor to suppress an adaptive immune response [35][116]. AhR activation is associated with TGF-β production, as well as IDO expression in dendritic cells, and it promotes differentiation of naive cluster of differentiation (CD) 4+ T-cells into immunosuppressive FoxP3+ regulatory T cells (Tregs) but not pro-inflammatory T helper (Th) 7 cells, through the AhR–Src–IDO1 pathway [36][117]. Activation of the KYN pathway suppresses effector T cells and induces Tregs, leading immune status to a tolerogenic state [37][118]. Increased KYN mediates inhibition of IL-2 signaling to reduce CD4+ T-cell survival [38][119]. IDO-expressing cells promote the differentiation of CD4+ T cells into Tregs expressing cytotoxic T-lymphocyte-associated protein 4, which downregulates immune responses [39][120]. Furthermore, higher KYNA production and lower KMO expression lead to dysfunctional effector CD4+ T-cell response [38][119]. NAD+ protects differentiated Th1, Th2, and Th17 from CD4+ T-cells, induces apoptosis of naïve CD4+ T-cells, and reduces the number of Tregs, but protects induced Tregs against apoptosis [40][121]. Thus, the KYN metabolic pathway enzymes and metabolites facilitate a shift toward tolerogenic T-cell functions. PA activates macrophages. PA potently induces production of macrophage inflammatory protein-1α and -1β, which contributes to tumor suppression [41][122]. Interacting synergistically with IFN-γ, PA increases inducible nitric oxide synthase messenger RNA (mRNA) expression in macrophages leading to potent cytotoxic and cytostatic actions. PA-treated macrophages inhibit tumor growth and increase survival in cancer animal models. PA is an endogenous metal chelator for iron which decreased proliferation rates of tumor cells in vitro and in vivo but did not affect normal human cells at the same dosage [42][123].2.3. Kynurenine Pathway Enzyme Activities
Inflammation activates several key enzymes in the KYN pathway. TDO in the liver, IDO 1 in the brain and peripheral tissues, and IDO 2 in the liver, kidney, and antigen-presenting cells (APCs) are the first rate-limiting enzymes of TRP metabolism [12][93]. The GC stress hormone, cortisol, activates TDO. The pro-inflammatory cytokines, IFN-α, IL-1β, IFN-γ, and TNF-α, activate IDO 1, while the anti-inflammatory cytokines, IL-2, IL-4, IL-10, and TGF-β, through IFN-γ, inhibit IDO 1 [12][93]. IDO 2 has a pro-inflammatory role, contributing to autoantibody production [14][95]. In addition, IDO-activated cells can alter the cytokine production of APCs to anti-inflammatory cytokines, TGF-β and IL−10, from pro-inflammatory cytokine, IL-12 [36][117]. The pro-inflammatory cytokine IFN-γ stimulates formamidase in human microglial cells and macrophages, leading to increased KYN synthesis. The activity of KATs is implicated in neurological and cognitive symptoms and the elderly [43][44][124,125]. A higher local KYN concentration is necessary for higher activity of KATs due to its low affinity. A cofactor, PLP, the active form of vitamin B6, and a cosubstrate, α-ketoacid, are required for KATs. [45][126]. A main source of PLP is food and degraded PLP-dependent enzymes by salvage pathway enzymes in humans. Genetic dysfunction of the salvage pathway enzymes and drug interactions of PLP or pyridoxal kinase result in convulsions and epileptic encephalopathy. A lower level of PLP is associated with neurological disorders including AD, PD, and epilepsy [46][47][127,128]. About 20% of the elderly are observed to have lower dietary vitamin B6 intake, and vitamin B6 supplementation improves cognitive performance in the elderly. It was proposed that folate, vitamin B6, and vitamin B12 are associated with cognitive performance [43][44][124,125]. The pro-inflammatory cytokine IFN-γ stimulates KMO activities in human microglial cells and macrophages, leading to increased QA synthesis. The activation of macrophages and glial cells induces the increased production of QA [48][129]. A lower KMO expression, in addition to higher KYNA production, is associated with dysfunctional effector CD4+ T-cell response, leading to suppression of adaptive immune response [49][130]. Microglial cells, which lack KATs but contain all enzymes that are involved in the successive conversion of KYN to QA, are believed to account for the local synthesis of 3-HK, 3-HAA, and QA. Moreover, microglia cells are responsible for the substantial upregulation of this major KP branch that is observed when the immune system is stimulated. KYNA synthesis, on the other hand, appears to occur almost exclusively in astrocytes, which lack KMO [50][131] (Figure 12).2.4. Systematic Reviews on Kynurenines in Major Neurodegenerative Diseases
A systematic review on KYNs in dementia (major neurocognitive disorders) was reported previously [14][95]. Complementing the researchtudy, the systematic review was conducted on the levels of KYNs in MS, ALS, HAND, and CJD. A total of 157 articles were found in the researchers'our database search. One systematic review and 22 articles were evaluated for eligibility. Finally, six articles were included in the source paper. The methodological quality and risk of bias assessment are presented in Table 1. Evidence levels of neurotoxic and neuromodulatory KYN levels were evaluated at high risk of bias for MS, ALS, and HAND, while no study was found in CJD.Table 1. Systematic synthesis of pro-inflammatory and anti-inflammatory cytokine levels in neurodegenerative diseases. ↑: increase; ↓: decrease; -: unchanged. HIV—human immunodeficiency virus.
| Diseases | Pro-Inflammatory Cytokines | Anti-Inflammatory Cytokines |
|---|---|---|
| Alzheimer’s disease | ↑ | ↑ |
| Parkinson’s disease | ↑ | ↑ |
| Multiple sclerosis | ↑ | ↓ |
| Huntington’s disease | ↑ | ↑ |
| Amyotrophic lateral sclerosis | ↑ | - |
| Creutzfeldt–Jakob disease | ↑ | ↑ |
Table 2. Systematic synthesis of neurotoxic and neuromodulatory kynurenine levels in neurodegenerative diseases. ↑: increase; ↓: decrease; ↑ ↓: mixed result; ?: unknown.
| Diseases | Neurotoxic Kynurenines | Neuromodulatory Kynurenines |
|---|---|---|
| Alzheimer’s disease | ↑ | ↓ |
| Parkinson’s disease | ↑ | ↓ |
| Multiple sclerosis | ↑ | ↑ ↓ |
| Huntington’s disease | ↑ | ↓ |
| HIV-associated neurocognitive disorders | ||
| ↑ | ↑ | |
| Stroke-induced secondary neurodegeneration | ↑ | ↓ |
| Amyotrophic lateral sclerosis | ||
| ↑ | ||
| ↑ ↓ | ||
| Creutzfeldt–Jakob disease | ? | ? |
| HIV-associated neurocognitive disorders | ↑ | ↑ |
| Stroke-induced secondary neurodegeneration | ↑ | ↑ |
2.5. Synthesis of Inflammatory Cytokines and Bioactive Kynurenines: Signs and Symptoms
Of the major NDs, motor dysfunctions are significant in PD, MS, HD, ALS, HAND, and SND, while autonomic dysfunctions are significant in MS, and relatively significant in PD, HD, ALS, CJD, HAND, and SND. Psychobehavioral symptoms are significant in AD, MS, CJD, and HAND and relatively significant in PD, HD, ALS, and SND. All NDs presented evidence of innate inflammatory activation by increased pro-inflammatory cytokines. AD, PD, HD, CJK, and HAND showed activation of the secondary adaptive immune response by increased anti-inflammatory cytokines. However, MS and SND showed reduced levels of anti-inflammatory cytokines. Regardless of the activation or inactivation of the secondary adaptive immune response, the inflammatory profiles of major NDs described in this resviearchw deviated from a healthy state. Either causative or resultant of acute and chronic inflammation, the altered balance of bioactive KYNs was observed. Neurotoxic KYNs are increased in all major NDs. Neuromodulatory KYNs were increased in HAND and SND, decreased in AD, PD, and HD, and remained mixed in MS and ALS (Figure 23).
Figure 23. The status of inflammatory cytokines and kynurenines: signs and symptoms of neurodegenerative diseases. All neurodegenerative diseases significantly increased pro-inflammatory cytokines and neurotoxic cytokines. The involvement of three representative symptoms of neurological diseases is described in lower axes of the profiles: motor dysfunction, autonomic dysfunction, and psychobehavioral symptoms.