More than 97% of patients with acute myeloid leukemia (AML) demonstrate genetic mutations leading to excessive proliferation combined with the evasion of regulated cell death (RCD). The most prominent and well-defined form of RCD is apoptosis, which serves as a defense mechanism against the emergence of cancer cells. Apoptosis is regulated in part by the BCL-2 family of pro- and anti-apoptotic proteins, whose balance can significantly determine cell survival. Apoptosis evasion plays a key role in tumorigenesis and drug resistance, and thus in the development and progression of AML. Research on the structural and biochemical aspects of apoptosis proteins and their regulators offers promise for new classes of targeted therapies and strategies for therapeutic intervention.
- acute myeloid leukemia
- apoptosis
- neddylation
1. Apoptosis
2. Pathways of Apoptosis
3. The Mechanism of the Mitochondrial Apoptosis Pathway
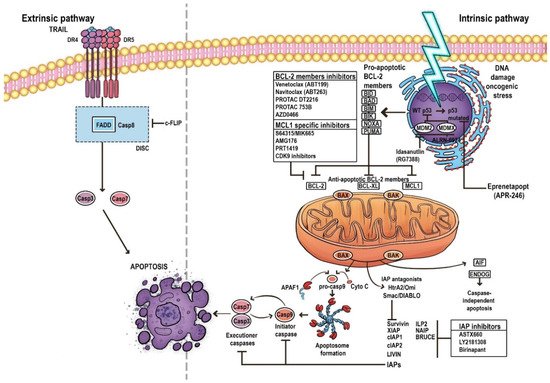
4. BCL-2 Family
5. Evasion of Apoptosis in AML
References
- Testa, U.; Riccioni, R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica 2007, 92, 81–94.
- Li, B.; Dou, S.-X.; Yuan, J.-W.; Liu, Y.-R.; Li, W.; Ye, F.; Wang, P.-Y.; Li, H. Intracellular transport is accelerated in early apoptotic cells. Proc. Natl. Acad. Sci. USA 2018, 115, 12118–12123.
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633.
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; D’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925.
- Letai, A. Apoptosis and Cancer. Annu. Rev. Cancer Biol. 2017, 1, 275–294.
- Koren, E.; Fuchs, Y. Modes of Regulated Cell Death in Cancer. Cancer Discov. 2021, 11, 245–265.
- MacKenzie, S.H.; Clark, A.C. Death by Caspase Dimerization. In Protein Dimerization and Oligomerization in Biology; Springer: Berlin/Heidelberg, Germany, 2012; Volume 747, pp. 55–73.
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193.
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120.
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta 2020, 1867, 118688.
- Hatok, J.; Racay, P. Bcl-2 family proteins: Master regulators of cell survival. Biomol. Concepts 2016, 7, 259–270.
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517.
- Zhang, L.-N.; Li, J.-Y.; Xu, W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther. 2012, 20, 1–7.
- Valentin, R.; Grabow, S.; Davids, M.S. The rise of apoptosis: Targeting apoptosis in hematologic malignancies. Blood 2018, 132, 1248–1264.
- Cassier, P.A.; Castets, M.; Belhabri, A.; Vey, N. Targeting apoptosis in acute myeloid leukaemia. Br. J. Cancer 2017, 117, 1089–1098.
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: P53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135.
- Kitada, S.; Pedersen, I.M.; Schimmer, A.D.; Reed, J.C. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene 2002, 21, 3459–3474.
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448.
- Hunter, A.M.; Sallman, D.A. Current status and new treatment approaches in TP53 mutated AML. Best Pract. Res. Clin. Haematol. 2019, 32, 134–144.
- Wang, C.; Sallman, D.A. What Are the Prospects for Treating TP53 Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia? Cancer J. 2022, 28, 51–61.
- Hou, H.-A.; Chou, W.-C.; Kuo, Y.-Y.; Liu, C.-Y.; Lin, L.-I.; Tseng, M.-H.; Chiang, Y.-C.; Liu, M.-C.; Liu, C.W.; Tang, J.-L.; et al. TP53 mutations in De Novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331.
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosom. Cancer 2019, 58, 875–888.
- Shallis, R.M.; Bewersdorf, J.P.; Stahl, M.F.; Halene, S.; Zeidan, A.M. Are We Moving the Needle for Patients with TP53-Mutated Acute Myeloid Leukemia? Cancers 2022, 14, 2434.
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096.
- Niparuck, P.; Police, P.; Noikongdee, P.; Siriputtanapong, K.; Limsuwanachot, N.; Rerkamnuaychoke, B.; Chuncharunee, S.; Siriboonpiputtana, T. TP53 mutation in newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Diagn. Pathol. 2021, 16, 100.
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. P53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2016, 31, 1296–1305.
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271.
- Jan, R.; Chaudhry, G.-E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218.
- Hrdinka, M.; Yabal, M. Inhibitor of apoptosis proteins in human health and disease. Genes Immun. 2019, 20, 641–650.
- Cheung, C.H.A.; Chang, Y.-C.; Lin, T.-Y.; Cheng, S.M.; Leung, E. Anti-apoptotic proteins in the autophagic world: An update on functions of XIAP, Survivin and BRUCE. J. Biomed. Sci. 2020, 27, 31.
- Oberoi-Khanuja, T.K.; Murali, A.; Rajalingam, K. IAPs on the move: Role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis. 2013, 4, e784.
- Pluta, A.; Wierzbowska, A.; Cebula-Obrzut, B.; Pluta, P.; Stępka, K.; Szmigielska-Kapłon, A.; Grzybowska-Izydorczyk, O.; Czemerska, M.; Smolewski, P.; Wrzesien-Kus, A.; et al. Prognostic value of inhibitor of apoptosis protein family expression in patients with acute myeloid leukemia. Leuk. Lymphoma 2015, 56, 2529–2535.
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.-M.; Büsselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919.
- Zhang, Y.; Huang, F.; Luo, Q.; Wu, X.; Liu, Z.; Chen, H.; Huang, Y. Inhibition of XIAP increases carboplatin sensitivity in ovarian cancer. Onco Targets Ther. 2018, 11, 8751–8759.
- Martinez-Ruiz, G.; Maldonado, V.; Ceballos-Cancino, G.; Grajeda, J.P.R.; Melendez-Zajgla, J. Role of Smac/DIABLO in cancer progression. J. Exp. Clin. Cancer Res. 2008, 27, 48.
- Mastrangelo, E.; Vachette, P.; Cossu, F.; Malvezzi, F.; Bolognesi, M.; Milani, M. The Activator of Apoptosis Smac-DIABLO Acts as a Tetramer in Solution. Biophys. J. 2015, 108, 714–723.