2. RT-Induced Cancer Cell Death
The main goal of RT is to destroy or slow tumor growth by using high-energy radiation, such as X-rays, gamma (γ) rays, electrons, protons, neutrons, and carbon ions. The efficacy of killing is influenced by a number of factors, including the type of radiation, the total dose, the fractionation rate, and the targeted organs. It is noted that different tumors show different levels of sensitivity to RT. Previous studies have shown that the primary intracellular target of RT is DNA
[5]. RT triggers DNA damage through the direct deposition of ionizing energy into the DNA or via the production of free radicals. Consequently, the damaged DNA is efficiently detected and repaired through homologous recombination (HR) or non-homologous end-joining (NHEJ) mechanisms. At present, NHEJ is the preferred pathway for repairing RT-induced DNA damage. A high radiation-associated deletion burden was associated with poor survival and may be able to predict the sensitivity of recurrent cancer after RT
[6]. Conversely, improper or inefficient mechanisms of DNA repair induce different manners of cell death, including mitotic catastrophe, apoptosis, and senescence (
Figure 1). Comprehensive reviews on the molecular mechanisms of these cell death phenotypes have previously been studied and published
[7][8][7,8].
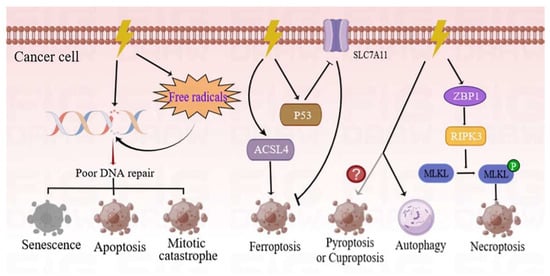
Figure 1. Different forms of cancer cell death programs induced by RT. RT can directly damage the DNA through production and deposition of ionizing energy or indirectly via free radicals. The DNA strands can be damaged in single-strand and double-strand breaks. If these DNA lesions cannot be properly repaired, tumor cells will initiate different types of death programs, including mitotic catastrophe, apoptosis, and senescence. Ionizing radiation (IR) can induce not only the expression of ACSL4 but also the activation of p53, and hence, results in elevated ferroptosis. The ZBP1-RIPK3-MLKL necroptotic cascade induces accumulation of cytoplasmic mtDNA in irradiated tumor cells, and consequently, activates an anti-tumor immune response. Conversely, autophagy is more inclined to cause cancer cell survival following IR treatment. However, there is a need for further studies to determine whether RT can induce pyroptosis or cuproptosis. ACSL4—acyl-CoA synthetase long-chain family member 4; p53—tumor protein p53; ZBP1—Z-DNA-binding protein 1; RIPK3—receptor-interacting serine/threonine kinase 3; MLKL—mixed-lineage kinase domain-like pseudokinase; mtDNA—mitochondrial DNA.
The modes of cancer cell death induced by RT in recent years are primarily summarized in the current
enst
rudy. Among them, ferroptosis is an iron-dependent cell death strategy which is triggered by excessive lipid peroxidation. Notably, RT can induce ferroptosis by inducing the expression of acyl-CoA synthetase long-chain family member 4 (ACSL4)
[9]. Simultaneously, the tumor protein p53 enhances RT-induced ferroptosis, partly by inhibiting the expression of solute carrier family 7 member 11 (SLC7A11)
[10]. As a new form of immunogenic cell death program, necroptosis, which is regulated by Z-DNA-binding protein 1 (ZBP1)-mixed-lineage kinase domain-like pseudokinase (MLKL) signaling, has been found to improve the radiation-induced antitumor immunity of cells
[11]. On the contrary, autophagy is more inclined to induce tumor resistance and is discussed in a subsequent section of this
enst
rudy. However, there is a need for further investigation to determine whether RT is able to induce pyroptosis or cuproptosis.
In general, there is a dose–effect relationship between the radiation dose and tumor control rate (that is, the increase in radiation dose can increase the killing effect of tumor cells within a certain scope). Most of
our
esearchers' understanding of radiobiological principles is based on conventional photon radiotherapy (doses of 1.8–2.0 Gy per day) outcome studies. The most widely accepted model is the linear–quadratic (LQ) model. Currently, RT has entered the era of unconventional fractionated radiotherapy, including stereotactic body radiotherapy (SBRT), and FLASH RT. While the LQ model has been widely used for decades in radiation oncology, its applicability to hypofractionated RT remains controversial. Additional efforts are required to fully exploit the potential of these novel RT techniques to guide clinical interventions.
3. Molecular Mechanisms of Radioresistance in Tumor Cells
Radioresistance is a major impediment to therapeutic efficacy and results in tumor recurrence, as well as distant metastasis. Previous studies have recently reported on the mechanisms of radioresistance mediated by the tumor stroma
[12][13][12,13]. The present
enst
rudy mainly reviews the radioresponse of resistant cancer cells (
Figure 2). Moreover, the post-radiation-exposure tumor-induced immunosuppression effects are also summarized in a subsequent section of the current
enst
rudy.
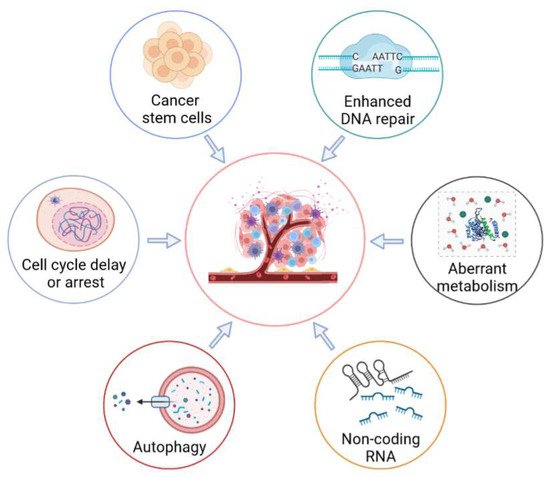
Figure 2. Molecular mechanisms of radioresistance mediated by tumor cells. Radioresistance of tumor cells is a major barrier to successful cancer treatment. In order to evade cell death, tumor cells have developed a variety of strategies, including enhancing DNA repair, activating cell-cycle checkpoints, regulating the self-renewal and differentiation of cancer stem cells, enhancing cellular metabolism, regulating the activities of autophagy, and regulating the expression of non-coding RNA.
3.1. Enhanced DNA-Repair Capability
RT kills tumor cells mainly by inducing double-strand DNA breaks. Therefore, an increased ability for DNA repair is strongly correlated with radioresistance in cancer cells. Serine proteinase inhibitor clade E member 2 (SERPINE2) promotes the repair potential of HR by activating the downstream repair protein RAD51
[14]. In a different study, the kinase ATM phosphorylates and stabilizes zinc finger E-box binding homeobox 1 (ZEB1). Then, ZEB1 binds to ubiquitin-specific peptidase 7 (USP7) and enhances its ability to deubiquitylate as well as stabilize the checkpoint kinase 1
[15]. In addition, pancreatic progenitor cell differentiation and proliferation factor (PPDPF), as well as Bromodomain-containing 4 (BRD4), can also enhance the repair capacity of tumor cells
[16][17][16,17].
The deletion of leucine-rich repeat-containing protein 31 (LRRC31) can enhance DNA repair through genome-wide CRISPR library screening. This occurs by through regulation of the activity of DNA-PKcs and ATR-MSH2 signaling
[18]. Previous studies have found that the repression of ubiquitin-conjugating enzyme E2O (UBE2O) or aurora kinase A (AURKA) can also promote radiation-induced DNA damage in lung cancer cells
[19][20][19,20].
3.2. Cell-Cycle Checkpoint Activation
The integrity of DNA molecular structure and function is essential in the maintenance of the normal cellular function. When the DNA structure is damaged, the cell cycle of the affected tissue can be delayed or arrested to provide sufficient time for DNA repair. Therefore, tumor cells can enhance resistance to radiation by regulating cell-cycle arrest. For instance, cyclin K plays pivotal roles in the regulation of the responses that lead to DNA damage and control the G2/M checkpoint through modulation of the β-catenin/cyclin D1 axis
[21]. A separate previous study has revealed that a positive feedback loop of the DNA-PK/AKT/GSK3β/cyclin D1 pathway can also activate the DNA-damage checkpoint
[22]. Ubiquitin-conjugating enzyme E2T (UBE2T) interacts with and monoubiquitinates H2AX upon exposure to radiation, and hence, facilitates the activation of CHK1, as well as the cell-cycle arrest
[23].
Topoisomerase IIβ-binding protein 1 (TopBP1) or Claspin is an important mediator in the checkpoint which increases the phosphorylation of CHK1 for the radioresistance of lung cancer cells
[24]. In addition, RT can drive p21-activated kinase 1 (PAK1) to phosphorylate RAF1 on serine 338 and recruit checkpoint kinase 2
[25]. Notably, the HPV oncoproteins E6 and E7 promote the expression of the Ras-associated binding protein Rab12 to facilitate the arrest of G2/M
[26]. Further, it has been shown that caspase-activated DNase (CAD) actively promotes self-inflicted DNA breaks, which leads to the arrest of the G2 phase in cancer cells, and hence, increases the survival of the cell after RT
[27].
3.3. Cancer Stem Cells (CSCs)
Cancer stem cells (CSCs) represent a small population of cells (approximately 1~2%) in tumor tissue with characteristics of stem cells. Increasing evidence indicates that CSCs are closely related to the occurrence, treatment, prognosis, recurrence, and metastasis of tumors
[28][29][30][28,29,30]. Fractionated irradiation promotes N-cadherin expression, which maintains the stemness of glioma stem cells by inhibiting Wnt/β-catenin signaling
[31]. On the contrary, ribosomal S6 protein kinase 4 (RSK4) promotes CSC properties in esophageal squamous cell carcinoma (ESCC) by activating the Wnt/β-catenin pathway
[32]. In colorectal cancer cells, JAK2/STAT3/CCND2 signaling contributes to cancer cell stemness and radioresistance
[33]. In addition, it has been found that S100 calcium-binding protein A9 (S100A9) mediates the properties of CSCs in the brain microenvironment by activating the RAGE/NF-κB pathway
[34]. It has been reported that with ultra-high doses of irradiation (~10
9 Gy/s), the CSCs may enhance lysosome-mediated autophagy and decrease pyroptosis and apoptosis, as well as necrosis
[35].
Pluripotency transcription factors, such as octamer-binding transcription factor 4 (OCT4), SRY-box transcription factor 2 (SOX2), and Nanog homeobox (NANOG), play an important role in the regulation of the self-renewal and differentiation of CSCs. It has been noted that THO complex 2/5 can facilitate the mRNA export and translation of SOX2 and NANOG
[36]. Similarly, RAD51AP1 regulates the self-renewal of CSCs in breast cancer cells
[37]. Additionally, the upregulation of ALG3 promotes the glycosylation of transforming growth factor beta receptor 2 (TGFBR2). This later activates the TGF-β/Smad pathway, which further promotes the expression of OCT4, NANOG, and SOX2
[38]. Moreover, the positive-feedback activation of ROS/AKT signaling contributes to the enrichment of CSCs in nasopharyngeal carcinoma
[39].
3.4. Aberrant Metabolism
Deregulating cellular metabolism is a hallmark of cancer, and hence, can regulate the resistance of cancer cells to therapy
[40]. For instance, the high expression of glutamine synthetase can promote nucleotide metabolism for the efficient repair of damaged DNA, and hence, contributes to the resistance of tumor cells against radiation
[41]. In lung cancer, KEAP1/NFE2L2 mutations depend on glutamine metabolism to decrease intracellular ROS levels
[42]. In addition, purine metabolism has been found to protect glioblastoma from radiation by promoting the repair of damaged DNA
[43]. It has also been reported that metabolic pathways which are regulated by nicotinamide adenine dinucleotide (NAD
+) and nicotinamide adenine dinucleotide phosphate (NADPH) can also potentiate radiation resistance in glioblastoma
[44][45][44,45]. Moreover, oncostatin M receptor (OSMR) promotes oxidative phosphorylation by interacting with the NADH ubiquinone oxidoreductase core subunit S1/2 (NDUFS1/2)
[46].
Recently, fatty acids have been found to be an alternative pivotal resource for energy, and thus, contribute to radiation-induced resistance by facilitating the oxidation of mitochondrial fatty acids (FAO)
[47][48][49][47,48,49]. Notably, fatty acid metabolism can also enhance CD47-mediated anti-phagocytosis in glioblastoma multiforme. Mechanistically, acetyl-CoA, which is one of the main metabolites of FAO, upregulates the expression of CD47 by acetylating RelA K310
[47]. It is also notable that the crosstalk between lipid droplets and iron metabolism makes cancer cells more radioresistant
[50]. In hepatocellular carcinoma (HCC) cells, the integration of glucose and cardiolipin anabolism, which is regulated by the mTORC1/HIF-1α/SREBP1 axis, promotes resistance to radiation by repressing cytochrome c extrusion
[51].
4. RT-Enhanced Tumor Metastasis
Tumor metastasis is the primary cause of failure in conventional cancer therapy
[39]. Currently, the biological responses of normal tissues to RT-induced damage and the influence of them on metastatic proficiency are not fully understood. A recent study indicates that radiation exposure in healthy lung tissue induces a hospitable environment for metastatic growth. Mechanistically, RT induces neutrophil accumulation and activation in healthy lung tissue, leading to a range of tissue perturbations, including Notch activation
[52][65]. Additionally, the epithelial–mesenchymal transition (EMT) is a biological process in which epithelial cells acquire mesenchymal characteristics, which makes them more invasive and metastatic. In cancer, this program is hijacked to endow cancer cells with tumor-initiating and metastatic potential
[53][54][66,67]. For instance, RT can promote the expression of ADAM metallopeptidase domain 10 (ADAM10) in pancreatic cancer cells, leading to the cleavage of ephrinB2, which is expressed in stromal fibroblasts. Subsequently, the extracellular domain of ephrinB2 interacts with its receptor and drives EMT and invasion
[55][68]. It has also been reported that RT can promote the stability of cell division cycle 6 (CDC6) protein, thereby promoting cell invasion and migration
[56][69]. Moreover, the epithelial cell adhesion molecule (EpCAM) is associated with a hybrid epithelial–mesenchymal phenotype in breast cancer
[57][70].