Responsible for tularemia, Francisella tularensis bacteria are highly infectious Gram-negative, category A bioterrorism agents. The molecular mechanisms for their virulence and resistance to antibiotics remain largely unknown. FupA (Fer Utilization Protein), a protein mediating high-affinity transport of ferrous iron across the outer membrane, is associated with both. Recent studies demonstrated that fupA deletion contributed to lower F. tularensis susceptibility towards fluoroquinolones, by increasing the production of outer membrane vesicles. Although the paralogous FupB protein lacks such activity, iron transport capacity and a role in membrane stability were reported for the FupA/B chimera, a protein found in some F. tularensis strains, including the live vaccine strain (LVS). To investigate the mode of action of these proteins, we purified recombinant FupA, FupB and FupA/B proteins expressed in Escherichia coli and incorporated them into mixed lipid bilayers. We examined the porin-forming activity of the FupA/B proteoliposomes using a fluorescent 8-aminonaphthalene-1,3,6-trisulfonic acid, disodium salt (ANTS) probe. Using electrophysiology on tethered bilayer lipid membranes, we confirmed that the FupA/B fusion protein exhibits pore-forming activity with large ionic conductance, a property shared with both FupA and FupB. This demonstration opens up new avenues for identifying functional genes, and novel therapeutic strategies against F. tularensis infections.
- Francisella tularensis
- fluorescence flux
- impedance spectroscopy
- FupA
- FupB
- porins
- Definition
1. Introduction
The facultative intracellular Gram-negative coccobacillus Francisella tularensis is the etiologic agent of tularemia [1]. The only two subspecies of F. tularensis that cause severe disease in humans are the subsp. tularensis (Type A strains) and subsp. holarctica (Type B strains). Because of its extremely high infectivity (<10 bacteria are sufficient to induce severe infection) and its potential use as a biological weapon, the aerosolizable and high-mortality rate Francisella pathogen has been classified as a class A bioterrorism agent by the U.S.A. Centers for Disease Control and Prevention (CDC).
- History
2. History
No safe and potent vaccine is currently available to prevent infection by
[
]. In the mid-1990s, Russian scientists developed a vaccine based on an attenuated mutant selected from a virulent isolate of
subsp.
[
]. This
LVS was used for protection of at-risk laboratory staff. However, because this strain was lethal in mice and its reversion to virulence could not be excluded, it remained unlicensed in the USA and the European Union [
]. A later study which aimed at the creation of an attenuated Type A mutant strain led to the discovery of a spontaneous
subsp.
SCHU S4 mutant, which was designated FSC043. It exhibited an improved efficacy against both systemic and aerosol challenges in mice [
]. Proteomic analysis of this mutant identified protein candidates that could be responsible for the reduction in virulence. Among these candidates were the paralogous proteins encoded by FTT0918 and FTT0919. Neither protein was expressed in the FSC043 strain, which instead codes for a hybrid protein that consists of the FTT0918 N-terminal and the FTT0919 C-terminal domains. Interestingly, such a fusion protein, which is believed to originate from a genomic deletion resulting from a recombination event between the two paralogous genes, was also observed in the LVS strain [
]. In contrast to the deletion of the gene encoding FTT0919 that had no effect on bacterial virulence in a mouse infection model, inactivation of the gene encoding FTT0918 was found to contribute significantly to the attenuation of both the SCHU S4 [
] and the LVS strains [
]. The name FupA (Fer Utilization Protein A) was proposed for this 58 kDa protein since it was very similar to the
siderophore receptor FslE that was required for the acquisition of siderophore-bound iron [
]. The name FupB was given to the protein encoded by the adjacent FTT0919 paralogous gene for which no relationships with metal acquisition or bacterial virulence had been shown [
,
]. The LVS chimeric protein (locus FTL0439) implicated in iron metabolism was termed FupA/B [
].
We recently performed a comprehensive phylogenetic analysis of the FupA and FupB homologous proteins [
]. These outer membrane proteins (OMPs) comprise the five subfamilies FupA, FupB, FslE, FmvA, and FmvB, which share a domain of unknown function (DUF3573) and display specific features that might reflect functional differences. To date, very little is known about the functions of these proteins. From the observation that FupA expression is regulated neither by the ferric uptake regulator (Fur) protein nor by the iron level, it was suggested that this protein may not be involved in iron acquisition and that it could instead be involved in the transport of additional substrates [
]. Among such unexpected functions, we demonstrated that the deletion of
or
contributes to reduced antibiotic susceptibility and could promote the emergence of antibiotic resistance mediated by an increased biofilm formation [
]. This effect was related to the hyper-production of vesicles by the
deletion mutant and correlates well with the presence of a lipoprotein motif at the N-terminus of FupA, which deletion would compromise the membrane stability. The functional role of FupB remains to be clarified since, in contrast to what had been described for
or
, removal of
failed to alter iron metabolism [
], bacterial virulence [
], or antibiotic resistance [
].
Although FupA and FupB are identified as orthologous proteins using the BLASTP software, their functions are not preserved [
,
]. The evolution of gene-phenotype relationships is complex, and functional differences between orthologues are, in some cases, greater than expected [
]. Due to the absence of structural data available in the proteomic databases, we first performed structural modeling using protein structure prediction programs in order to gain some insight into the functional roles of both FupA and FupB. Bioinformatic simulations provided evidence that, as previously predicted for FupA using the Hidden Markov Model-based PRED-TMBB program [
], both FupB and FupA/B fold partially as β-barrels in the outer membrane of
. FupA and FupA/B have been reported to facilitate iron transport [
,
,
].
-
Results and Discussion
3. Results and Discussion
3.1. Structural Modelling of Fup Proteins Predicted Their Folding as Porins
FupA and FupB are paralogous proteins that consist in 558 and 482 amino acids, respectively. Alignment of both proteins with the 552 amino acid sequence of FupA/B has already been reported [
]. The amino acids 1 to 307 of FupA/B are 98.7% identical to the N-terminal region of FupA, while its C-terminal half (amino acids 298 to 552) is strictly identical to the C-terminal part of FupB. The three-dimensional (3D) structures of the three proteins were predicted using the Phyre 2 server [
]. The automatic alignments of the C terminal regions of FupA and FupA/B (56% of their sequence) and FupB (63% of its sequence) with available structures were modeled with respectively 98.3% (FupA), 98.9% (FupB), and 98.8% (FupA/B) accuracies by the single highest scoring template. As expected when considering amino acid sequence homologies, the three partial predicted structures (
) were very similar to each other and showed clear homologies with known porins mainly folded as β-barrels, in particular with the outer membrane protein OprP from
[
]. The obtained model (
) depicted the
Fup proteins as monomers spanning the membranes by the arrangement of a 16-stranded β-barrel. As for OprP, FupA, FupB, and FupA/B are expected to form trimers.
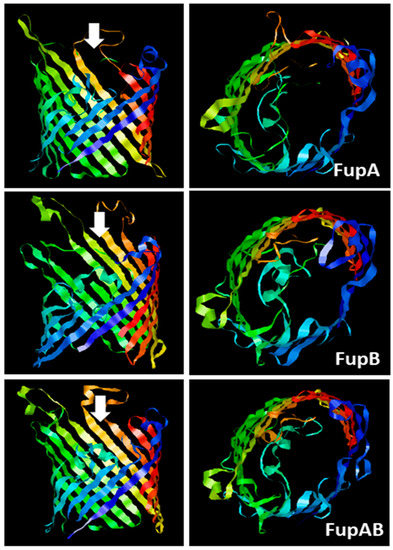
Predicted 3D structures of
FupA, FupB, and FupA/B monomers. The C terminal regions of the 3 proteins (310 amino acids from C terminal part of FupA; 304 amino acids C terminal part of FupA/B and FupB) were analyzed using the Phyre2 server. The panels on the left show a lateral view of each transmembrane region constituted of 16-barrels, while the panels on the right show a top view of each transmembrane region, observed from the intracellular compartment and in the direction of the white arrows.
3.2. Fluorescence Assays on Proteoliposomes Containing FupA/B Suggested Porin Activity
Insertion of the chimeric FupA/B protein into liposomes was used as a means to evaluate the insertion capability of the FupA protein family into membranes and their permeability to a fluorescent molecule. The integration of FupA/B into liposomes was analyzed through a discontinuous sucrose gradient. Dot blot analysis of the gradient fractions with anti-His antibodies failed to reveal the presence of FupA/B in the lowest fraction of the gradient (40% sucrose), indicating that the protein did not precipitate during the integration of the protein into the liposomes (
B). A thin white band associated with the formed PLs could be observed at the interface between the 0 and the 10% sucrose cushions (
B).
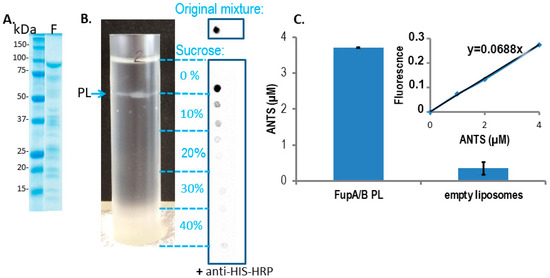
FupA/B inserts spontaneously into liposome lipid bilayers. (
) Coomassie blue staining gel of the purified MBP-FupA/B protein. Marker molecular weights are indicated (kDa). (
) The sucrose density gradient (0 to 40%) (left panel) and dot blot analysis of FupA/B (right panel) revealed that the PLs concentrated at the interface between the 0% and the 10% sucrose steps. 2 µL of each sucrose gradient fraction were spotted on a nitrocellulose membrane revealed by anti-His antibodies conjugated to horseradish peroxidase (HRP) and chemiluminescence. The top panel shows the signal obtained with 2 μg of recombinant FupA/B. (
) Comparison of the ANTS fluorescence signals (± SD) from 100 µL of FupA/B PL vs 100 µL of liposomes after G25 filtration (Ex: 405 nm; Em: 535 nm;
-value = 0.0014 between both histograms, non-parametric one-way ANOVA). A linear correlation between the ANTS concentration and the emitted fluorescence was verified via the titration of pure ANTS. These data are representative of 3 independent experiments performed in duplicate and providing similar results.
The number of FupA/B proteins per PL was calculated in several steps, as follows.
-
The intensity of the FupA/B light signal emitted by the PL dot was quantified as being 73% of that emitted by the dot corresponding to the starting material (FupA/B-MBP-His tagged protein at 100 μg/mL) (
B). Given that the sucrose gradient fractions were of 1 mL, we estimated that the whole PL fraction contained 73 μg of FupA/B protein. Knowing that the molar mass of MBP-FupA/B is 100 kDa (Reference [
] and
A), we estimated that there were 73 × 10
−11moles of FupA/B, i.e., 4.38 × 10
14molecules, of FupA/B inserted in the total number of PLs.
-
We calculated the number of lipid molecules per PL by dividing the whole surface occupied by lipids in one PL by the mean surface of one phospholipid. Dynamic Light Scattering (DLS) measured the mean radius of PLs as being 63.5 ± 3.6 nm (mean ± SD;
n= 3). Given that PLs contain 2 layers of lipids, we calculated the whole surface occupied by lipids in one PL as 2 × (4π R
2), thus as 1.01 × 10
5nm
2
]. Fluorescence measurement of the fraction collected from the G25 column, on which empty liposomes pre-incubated with ANTS had been loaded, revealed values close to 0. This indicated that the membrane of empty liposomes was not permeable to the ANTS dye and that filtration through the G25 column efficiently excluded ANTS from the liposome solution (
C, “empty liposomes” column). In contrast, measurement of the fluorescence of the fraction collected from the G25 column loaded with a mixture of PLs pre-incubated with ANTS returned an ANTS concentration of 3.70 ± 0.01 μM (
C, “FupA/B PL” column). Altogether, these results demonstrated that ANTS could penetrate FupA/B PL and that FupA/B is a porin that allows fluorescent molecules such as ANTS to pass through a membrane bilayer.
To calculate the amount of ANTS which had entered the PLs through the FupA/B porin, we proceeded again by steps, as follows:
-
Knowing that the PLs’ radius estimated by DLS was 63.5 ± 3.6 nm, we calculated the volume of any given PL as 4/3 × π × R
3= 1.072 × 10
−21m
3= 1.072 × 10
−18L.
. Knowing that the mean surface of a phospholipid is estimated to be 0.65 nm
2[
], we estimated the number of lipid molecules per PL as being 1.01 × 10
5/0.65 = 1.55 × 10
5.
-
Knowing that 2 mg of lipids had been used for the experiment, with the production of 1.013 × 10
13PLs, the total volume occupied by the formed PLs in the 1 mL working fraction was calculated as 1.013 × 10
13× 1.072 × 10
−18= 1.04 × 10
−5L = 11.04 μL. The total volume of PLs thus represented 1.10% of the 1 mL working fraction.
To calculate the number of formed PLs, we divided the total number of lipid molecules found in all the PLs by the number of lipid molecules per PL. The liposomes were prepared with 2 mg, thus 2.85 µM, i.e., 1.71 × 10
18molecules of lipids. Assuming that there was no loss of lipids between the liposome formation step and that of PLs, the number of formed PLs was thus estimated to be 1.71 × 10
18/1.55 × 10
5= 1.03 × 10
13.
-
Finally, the number of FupA/B monomers per PL was calculated as the total number of FupA/B proteins in the whole number of PLs, divided by the estimated number of PLs, thus 4.38 × 10
14/1.03 × 10
13= 39.7.
This number of protein monomers per PL was in the range of the typical membrane protein occupation range in PLs obtained using detergents [
].
To assess the potential porin activity of FupA/B, we then measured the efficacy of entrance of the ANTS dye into the PLs. Gel filtration through a Sephadex column had been reported as the method of choice for separating liposome-encapsulated molecules from those remaining outside the liposome membranes [
,
Given that PLs were incubated in a 20 mM ANTS solution and that the total volume of PLs represented 1.10% of the total volume (1 mL), we reasoned that the maximum concentration of ANTS reached inside the PLs would have been 20 × 10
- 3
× 1.10% = 220 μM if their membrane had been permeable to the dye. In contrast, we obtained a concentration of 3.70 ± 0.01 μM ANTS within the PLs (
C), which represented only 1.7% of the ANTS molecules that would have diffused passively through a permeable membrane.
-
Given the ANTS concentration calculated within the PLs (3.70 ± 0.01 µM) and the calculated volume of one PL (1.072 × 10
−18L), we calculated that each PL would contain 4.02 × 10
−24moles of ANTS, i.e., 2.42 molecules of ANTS, while it would have contained 142 molecules if the PL membrane had been permeable to ANTS.
This calculation clearly demonstrated that ANTS molecules did not diffuse passively through the empty PL membrane and that their entry into the PL required instead their fluorescent flux through the FupA/B proteins. This property strongly suggested a FupA/B porin activity.
3.3. Impedance Spectroscopy Experiments Using Tethered Lipid Bilayer Membranes Confirmed the Porin Activity of FupA/B
We have already shown in a previous study that impedance spectroscopy provides a precise measurement of the ion flux driven by the OprF porin across membranes [
]. To evaluate the porin activity of FupA, FupB, and FupA/B proteins, we thus used tethered lipid bilayer membranes (t-LBMs) generated by a TethaPod device (SDX Tethered Membranes, Australia). We first measured the steady-state conductance of this t-LBM system using impedance spectroscopy controlled by the TethaPod, in the presence of recombinant FupA/B vs buffer A (control experiment) added to one side of the t-LBM. The insertion of FupA/B into the t-LBM led to a reproducible and steady-state conductance that stabilized at 0.2–0.25 µS within a few minutes (
A). Specifically, we verified that the capacitance varied only slightly (18%) upon the incorporation of FupA/B to the t-LBM (
B). According to the manufacturer’s recommendations, an almost constant capacitance of ~10 nF upon addition of the protein to the t-LBM indicated that the thickness of the membrane did not vary through the experiment and thus excluded the possibility of a lipid multilayer, which would have been inappropriate for protein insertion into the membrane.
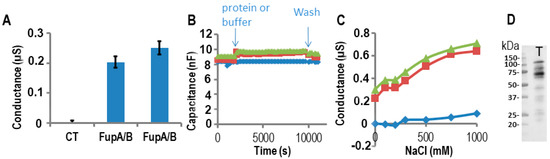
Measurements of FupA/B porin activity using impedance spectrometry on a TethaPod chip. FupA/B was digested by the TEV enzyme in the well. (
) The conductance of the t-LBM after the insertion of FupA/B (2 independent wells) and no protein (CT) in 50 mM Tris pH 7.5, 200 mM KCl. The conductances are the mean (± SD) of the conductances measured over 10 min. (
) The capacitance of the t-LBM before and after incorporation of FupA/B (2 independent wells: red squares and green triangles) or buffer (blue diamonds). The times of protein addition and washes are indicated. Fifteen data points were used for each measurement. (
) The conductance after NaCl addition in the upper compartment of the t-LBM after incorporation of FupA/B (2 independent wells: red squares and green triangles) or buffer (blue diamonds). The conductances are the mean of the conductances measured during NaCl incubation (10 min for each NaCl concentration). (
) Immunoblot analysis using the anti-FupA antibody of FupA/B solubilized in the t-LBM well by SDS (lane T). Marker molecular weights are indicated (kDa).
To characterize the ion permeability of the FupA/B porin, we added increasing NaCl concentrations into the upper part of the t-LBM wells. From
C, we concluded that the porin FupA/B was permeable to NaCl. In the control well (no protein) we noticed a small increase in conductance for concentrations of NaCl greater than 500 mM, which suggested that only a small increase in permeability of the t-LBM resulted from the very large osmotic difference across the membrane (in blue in
C).
Following conductance measurements, the proteins potentially inserted in the t-LBM were then solubilized by the addition of an SDS solution into the wells followed by their separation by SDS-PAGE and immunoblotting using antibodies specific to FupA. A band migrating at the apparent molecular weight expected for FupA/B confirmed the correct insertion of the protein in the t-LBM (
D, T lane). The size of FupA/B protein in lane T (~60 kDa,
D) is ~40 kDa less than that of the recombinant FupA/B detected by Coomassie blue staining of the SDS-PAGE shown in
A due to the TEV protease digestion. We concluded that the increase in conductance detected in
A was due to the porin activity of FupA/B inserted in the t-LBM.
Since FupA/B, FupA, and FupB are orthologous proteins, we tested the three proteins for their potential porin activities on the same Tethapod chip. In
A, we verified after washes of the well that the resulting conductance of FupA or FupB insertion in the t-LBM membrane was similar (0.2 µS) to the one obtained in
A for FupA/B. The washes of the well of the TethaPod did again produce a loss of conductance for FupA/B. The final conductances of FupA and FupB were around 0.25 and 0.3 µS, respectively (
B). The conductances of the 3 tested Fup proteins with or without their MBP tag were very similar. These high conductances suggested that the insertion of these proteins into t-LBM was effective. Their insertion in the t-LBM was as fast as the one of FupA/B. These experiments were done thrice on 3 different chips and we always found slightly more conductance with FupB than with FupA (around a 1.25-fold increase) (
B).
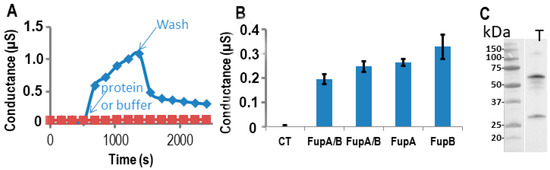
Measurements of Fup protein porin activity using impedance spectrometry. The proteins were digested by the TEV enzyme in the well. (
) The conductance of the t-LBM after the insertion of FupA/B (blue diamonds) and without protein (red squares). Time of protein (or buffer) addition and washes are indicated. (
) Conductance of the t-LBM (± SD) after the insertion of FupA or FupB compared to FupA/B (in two independent wells), and no protein (CT). The conductances are the mean of the conductances measured over 10 min. (
) Immunoblot analysis of FupA solubilized in the t-LBM well by SDS and revealed by antibodies specific to FupA (laneT). Marker molecular weights are indicated (kDa).
Finally, solubilization of the proteins present in the t-LBM by SDS followed by their separation by SDS-PAGE and immunoblotting using antibodies specific to FupA confirmed the insertion of FupA in the t-LBM and the apparent molecular mass detected for FupA was in agreement with its cleavage by the TEV protease (
C). Altogether, these experiments let us to conclude that FupA and FupB are porins.
- Oyston, P.C.F.; Sjostedt, A.; Titball, R.W. Tularaemia: Bioterrorism defence renews interest in Francisella tularensis. Rev. Microbiol 2004, 2, 967–978.
- Eigelsbach, H.T.; Downs, C.M. Prophylactic effectiveness of live and killed tularemia vaccines. I. Production of vaccine and evaluation in the white mouse and guinea pig. Immunol. 1961, 87, 415–425.
- Oyston, P.C.F. Francisella tularensis vaccines. Vaccine 2009, 27 (Suppl. S4), D48–D51.
- Twine, S.; Byström, M.; Chen, W.; Forsman, M.; Golovliov, I.; Johansson, A.; Kelly, J.; Lindgren, H.; Svensson, K.; Zingmark, C.; et al. A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Immun. 2005, 73, 8345–8352.
- Salomonsson, E.; Kuoppa, K.; Forslund, A.L.; Zingmark, C.; Golovliov, I.; Sjöstedt, A.; Noppa, L.; Forsberg, Å. Reintroduction of two deleted virulence loci restores full virulence to the live vaccine strain of Francisella tularensis. Immun. 2009, 77, 3424–3431.
- Lindgren, H.; Honn, M.; Golovlev, I.; Kadzhaev, K.; Conlan, W.; Sjöstedt, A. The 58-kilodalton major virulence factor of Francisella tularensis is required for efficient utilization of iron. Immun. 2009, 77, 4429–4436.
- Ramakrishnan, G.; Sen, B.; Johnson, R. Paralogous outer membrane proteins mediate uptake of different forms of iron and synergistically govern virulence in Francisella tularensis tularensis. Biol. Chem. 2012, 287, 25191–25202.
- Sen, B.; Meeker, A.; Ramakrishnan, G. The fslE homolog, FTL-0439 (fupA/B), mediates siderophore-dependent iron uptake in Francisella tularensis LVS. Immun. 2010, 78, 4276–4285.
- Siebert, C.; Lindgren, H.; Ferré, S.; Villers, C.; Boisset, S.; Perard, J.; Sjöstedt, A.; Maurin, M.; Brochier-Armanet, C.; Couté, Y.; et al. Francisella tularensis: FupA mutation contributes to fluoroquinolone resistance by increasing vesicle secretion and biofilm formation. Microbes Infect. 2019, 8, 808–822.
- Ramakrishnan, G. Iron and Virulence in Francisella tularensis. Cell. Infect. Microbiol. 2017, 7, 107.
- BLAST Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins (accessed on).
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Rev. Genet. 2013, 14, 360–366.
- Ramakrishnan, G.; Sen, B. The FupA/B protein uniquely facilitates transport of ferrous iron and siderophore-associated ferric iron across the outer membrane of Francisella tularensis live vaccine strain. Microbiology 2014, 160, 446–457.
- Pérez, N.; Johnson, R.; Sen, B.; Ramakrishnan, G. Two parallel pathways for ferric and ferrous iron acquisition support growth and virulence of the intracellular pathogen Francisella tularensis Schu S4. Microbiologyopen 2016, 5, 453–468.
- Henderson, J.C.; Zimmerman, S.M.; Crofts, A.A.; Boll, J.M.; Kuhns, L.G.; Herrera, C.M.; Trent, M.S. The Power of Asymmetry: Architecture and Assembly of the Gram-Negative Outer Membrane Lipid Bilayer. Rev. Microbiol. 2016, 70, 255–278.
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Protoc. 2015, 10, 845–858.
- Pongprayoon, P.; Beckstein, O.; Wee, C.L.; Sansom, M.S.P. Simulations of anion transport through OprP reveal the molecular basis for high affinity and selectivity for phosphate. Natl. Acad. Sci. USA 2009, 106, 21614–21618.
- Cortes, S.; Barette, C.; Beroud, R.; De Waard, M.; Schaack, B. Functional characterization of cell-free expressed Kv1.3 channel using a voltage-sensitive fluorescent dye. Protein Expr. Purif. 2018, 145, 94–99.
- Crouch, C.H.; Bost, M.H.; Kim, T.H.; Green, B.M.; Arbuckle, D.S.; Grossman, C.H.; Howard, K.P. Optimization of detergent-mediated reconstitution of influenza A M2 protein into proteoliposomes. Membranes 2018, 8, 103.
- Ruysschaert, T.; Marque, A.; Duteyrat, J.L.; Lesieur, S.; Winterhalter, M.; Fournier, D. Liposome retention in size exclusion chromatography. BMC Biotechnol. 2005, 5, 1–13.
- Vemuri, S.; Rhodes, C.T. Separation of liposomes by a gel filtration chromatographic technique: a preliminary evaluation. Acta Helv. 1994, 69, 107–113.
- Maccarini, M.; Gayet, L.; Alcaraz, J.P.; Liguori, L.; Stidder, B.; Watkins, E.B.; Lenormand, J.L.; Martin, D.K. Functional Characterization of Cell-Free Expressed OprF Porin from Pseudomonas aeruginosa Stably Incorporated in Tethered Lipid Bilayers. Langmuir 2017, 33, 9988–9996.
- Geertsma, E.R.; Nik Mahmood, N.A.B.; Schuurman-Wolters, G.K.; Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Protoc. 2008, 3, 256.
- Rigaud, J.-L. Membrane proteins: functional and structural studies using reconstituted proteoliposomes and 2-D crystals. J. Med. Biol. Res. 2002, 35, 753–766.
- Stockbridge, R.B.; Tsai, M.F. Lipid reconstitution and recording of recombinant ion channels. In Methods in Enzymology; 2015.
- Subrini, O.; Sotomayor-Pérez, A.C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a membrane-active peptide from the bordetella pertussis CyaA toxin. Biol. Chem. 2013, 288, 32585–32598.
- Cranfield, C.G.; Cornell, B.A.; Grage, S.L.; Duckworth, P.; Carne, S.; Ulrich, A.S.; Martinac, B. Transient potential gradients and impedance measures of tethered bilayer lipid membranes: Pore-forming peptide insertion and the effect of electroporation. Biophys J. 2014, 106, 182–189.