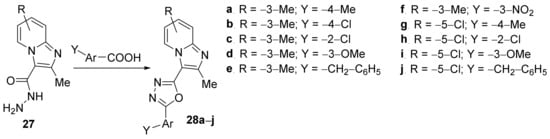2. Six-Member Ring Azaheterocycles with One Nitrogen Atom. Hybrid Pyridine
In their attempt to identify new antimicrobial compounds, Eryılmaz et al.
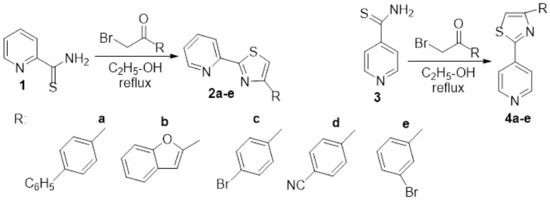
[9] designed and synthesized different hybrid pyridine derivatives bearing in the 2- and 4-position of the ring of a thiazole moiety. The synthesis was straight and efficient, involving a Hantzsch cyclocondensation of pyridine-2- and 4- carbothioamide
1 and
3 with acetophenone derivatives, when the desired hybrid 4-(
R-2-yl)-2-(pyridin-2-yl)thiazole
2a–
e and 4-(
R-2-yl)-2-(pyridin-4-yl)thiazole
4a–
e are obtained,
Scheme 1. The synthesized compounds were tested for their antibacterial activity [four strains,
Gram-positive (
Bacillus cereus,
Staphylococcus aureus) and
Gram-negative (
Escherichia coli,
Pseudomonas aeruginosa)] and antifungal activity (one strain,
Candida albicans)
via minimal inhibitory concentration (MIC) method and DNA cleavage activity studies. The researchers established interesting correlation structure-biological activity (SAR), the most relevant finding being that 4-pyridine thiazole hybrid compounds
4a–
e showed more potent activity than
2a–
e. The most promising compound was found to be
4c (MIC values 0.01 mM) exhibited on the bacterial strains
Staphylococcus aureus and
Bacillus cereus.
Scheme 1. Reaction pathway to obtain hybrid thiazole-pyridine 2a–e and 4a–e.
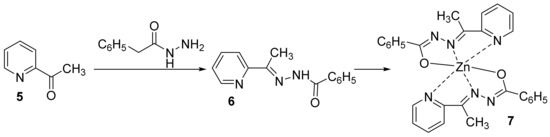
In a subsequent paper, some of the above researchers (Cinarli et al.
[10]) synthesized different hybrid aroylhydrazone-pyridine-metal derivatives. The newly hybrid aroylhydrazone-pyridine metal derivatives [ZnL
2]
7 have been synthesized in two steps: an initial cyclocondensation of pyridine-2-acyl derivative
5 (with aroylhydrazone leading to pyridine-aroylhydrazone ligand
6) is followed by complexation with M
2+ metal (Zn
2+),
Scheme 2.
Scheme 2. Reaction pathways to obtain hybrid metal-pyridine derivatives 7.
The synthesized compounds were tested for their antibacterial activity (four strains,
Pseudomonas aeruginosa,
Escherichia coli,
Bacillus cereus and
Staphylococcus aureus) and antifungal (one strain,
Candida albicans) activity
via minimal inhibitory concentration method. The [ZnL
2]
7 has been found to be more active than pyridine-aroylhydrazone ligand
6 in all microorganisms (MIC = 11.71 μg/mL for bacteria and MIC = 23.43 μg/mL for
C. albicans). The researchers claim that the synthesized new complex acts on microorganisms by disrupting the cell wall structure. The DNA binding interactions was also determined experimentally by spectrophotometric and electrochemical methods. The obtaining data indicate that ligand
6 and hybrid [ZnL
2]
7 interact the most with guanine base, and charge transfer is from DNA guanine bases to the molecular structures. Moreover, antioxidant activity was determined, and the hybrid [ZnL
2]
7 acted as a scavenger against peroxide radicals.
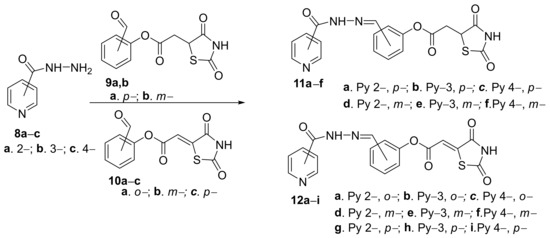
Trotsko et al.
[11] designed and synthesized different hybrid pyridine derivatives bearing at the 2-, 3- or 4- position of the ring of a thiazolidine-2,4-dione moiety. The synthesis involve a condensation reaction of hydrazonyl-pyridine
8a–
c with the corresponding (2,4-dioxo-1,3-thiazolidin-5-yl/ylidene)
9a,
b/
10a–
c, which are leading to the desired hybrid pyridine-2,4-dioxo-1,3-thiazolidin-5-yl derivatives
11a–
f or pyridine-2,4-dioxo-1,3-thiazolidin-5-ylidene derivatives
12a–
i,
Scheme 3.
Scheme 3. Reaction pathway to obtain hybrid thiazolidine-pyridine 11a–f and 12a–i.
The in vitro antimycobacterial assay (
Mycobacterium tuberculosis) of the newly obtained compounds reveals strong activity in the concentration range of 1–512 μg/mL and low cytotoxicity. Interesting SAR correlations have been performed, and the highest antimycobacterial activity (MIC = 1 μg/mL) was demonstrated for the hybrid pyridine derivatives bearing the thiazolidine-2,4-dione moiety at the 4-position of the pyridine ring (hybrids
11a–
c and
12g–
i).
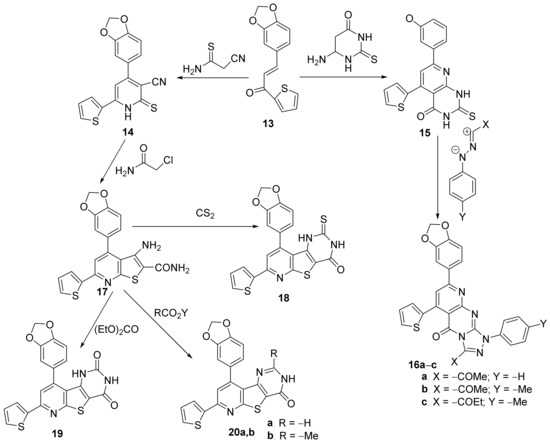
Sanad et al.
[12] have performed an interesting study concerning the in vitro antimicrobial activity of some newly hybrid thieno-pyrimidin-pyridine derivatives. The synthesized compounds belonged to different classes of substituted pyridine: thiophen-dihydropyridine
14, thiophen-pyrido-pyrimidin-4(1
H)-one
15, and fused pyridine: pyrido-thiophen-triazolo-pyrimidine
16a–
c, thiophen-pyrido-thieno derivative
17, thiophen-pyrido-thieno-pyrimidin-4-one
18, thiophen-pyrido-thieno-pyrimidin-2,4-dione
19, thiophen-pyrido-thieno-pyrimidin-2-
R-4-one
20,
Scheme 4.
Scheme 4. Reaction pathway to obtain hybrid thiophen-pyrimidin-pyridine 14–20.
The synthetic approach is straight and efficient, involving typical organic chemistry reactions, mostly cyclocondensations. The synthesized compounds were tested in vitro for their antibacterial activity against
Escherichia coli and
Klebsiella pneumoniae as
Gram-negative bacterial strains as well as against
Staphylococcus aureus and
Streptococcus mutans as
Gram-positive bacterial strains. The obtained results (expressed as the diameter of inhibition zones (DIZ) and MIC) reveal that the thiophen-pyrido-thieno-pyrimidin-2-
R-4-one
20a,
b exhibit the strongest antibacterial activities against all the tested bacteria, in the range of 40–60 mm for inhibition zones, respectively, 4–16 μg/mL for MIC values.
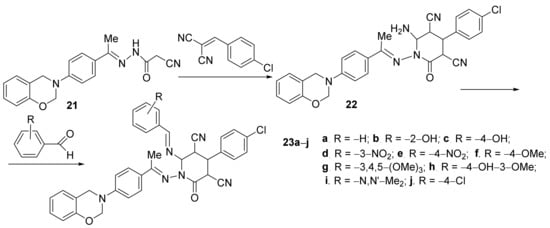
Desai et al.
[13] have studied the in vitro antimicrobial activities of some newly hybrid oxazino-pyridine derivatives. The desired compounds, oxazin-3(4
H)-yl)phenyl)ethyildene)amino)-6-((arylidene)amino)-4-(4-chlorophenyl)-2-oxo-1,2-dihydropyridine
23a–
j, were synthesized in two steps, by cyclocondensation of oxazine
21 followed by condensation of the intermediate
22,
Scheme 5.
Scheme 5. Reaction pathway to obtain hybrid oxazino-pyridine 23a–j.
The synthesized hybrid compounds were tested for their in vitro antibacterial activity against various bacteria (
Escherichia coli,
Pseudomonas aeruginosa,
Staphylococcus aureus,
Streptococcus pyogenes) and fungus (
Candida albicans,
Aspergillus niger,
Aspergillus clavatus)
via the MIC method. Some compounds have proved to have a very powerful activity against bacteria
E. coli (
23h, MIC = 25 μg/mL) and against fungus
C. albicans (
23f, MIC = 50 μg/mL), respectively,
A. clavatus (
23h, MIC = 25 μg/mL).
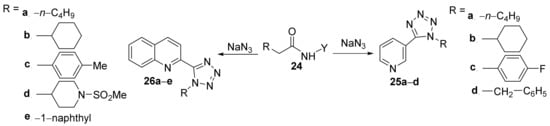
Sribalan et al.
[14] have studied thein vitroantimicrobial activity of some tetrazole-heterocycle hybrid derivatives. The synthesis supposes a cyclocondensation reaction of amide precursors
24 with sodium azide, when the corresponding tetrazolo-pyridine
25a–
d and tetrazolo-quinoline
26a–
e hybrids are obtained,
Scheme 6.
Scheme 6. Reaction pathway to obtain the tetrazolo-pyridine 25a–d and tetrazolo-quinoline 26a–e hybrids.
The synthesized tetrazolo-pyridine
25a–
d and tetrazolo-quinoline
26a–
e hybrids were tested for their in vitro antibacterial activity against various bacteria (
Klebsiella pneumoniae,
Pseudomonas aeruginosa,
Staphylococcus aureus,
Streptococcus pyogenes) and the fungus
Candida albicans. An interesting SAR correlation has been performed. The compound
25a (the pyridyl ring is decorated with
n-butyl) proved to be the most active from the tetrazolo-pyridine series against all bacteria (DIZ in the range of 4–15 mm), having a superior inhibition to the standard drug (amikacin). The compound
26d (the quinoline ring is decorated with a piperidyl-sulfonamide moiety) proved to be the most active from the tetrazolo-quinoline series against all bacteria (DIZ in the range of 4–10 mm), having a comparable inhibition to the standard. The antifungal activity was negligible.
Kuthyala et al.
[15] have studied the in vitro antimicrobial activity of some oxadiazolo-imidazopyridine hybrid derivatives. The synthesis was straight, involving a cyclocondensation reaction of hydrazonyl-imidazopyridine
27 with different benzoic acids, when the corresponding oxadiazolo-imidazopyridine hybrids
28a–
j were obtained,
Scheme 7.
Scheme 7. Reaction pathway to obtain oxadiazolo-imidazopyridine hybrids 28a–j.
The synthesized oxadiazolo-imidazopyridine hybrids
28a–
j were tested for their in vitro antibacterial activity against various human bacterial pathogens (
Escherichia coli,
Klebsiella pneumoniae,
Staphylococcus aureus,
Bacillus subtilis) and the fungus
Candida albicans and
Aspergillus niger. An interesting SAR correlation has been performed. The compounds
28f and
28g have high activity against
Gram-positive bacteria
S. aureus (MIC = 3.12 μg/mL), while compound
28f proved to have high activity against fungus
C. albicans (MIC = 12.5 μg/mL).
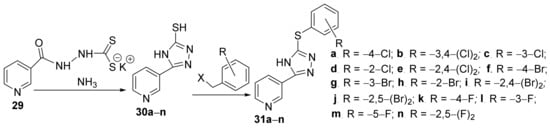
Ahirwaret al.
[16] synthesized two new series of some 1,3,4-triazolo-pyridine hybrid derivatives and studied their antimicrobial activities. The synthesis was conducted in two steps: a cyclocondensation reaction of dithiocarbazate
29 with ammonia leading to the first class of hybrids triazolo-pyridine
30a–
n, then an alkylation reaction of
30a–
n with benzyl halide takes place leading to the second class of hybrids triazolo-pyridine
31a–
n,
Scheme 8.
Scheme 8. Reaction pathway to obtain 1,3,4-triazolo-pyridine hybryds 30a–m and 31a–m.
The synthesized triazolo-pyridine hybrids
30a–
n and
31a–
n were evaluated for their in vitro antibacterial activity against
Gram-positive bacteria (three strains:
Staphylococcus aureus,
Streptococcus pyogenes,
Enterococcus faecalis) and
Gram-negative bacteria (three strains:
Escherichia coli,
Pseudomonas aeruginosa,
Acinetbacter baumannii) by MIC assay. From the tested compounds, two of them,
31h and
31i, have excellent activity against all strains (MIC in the range of 0.91–11 μg/mL).
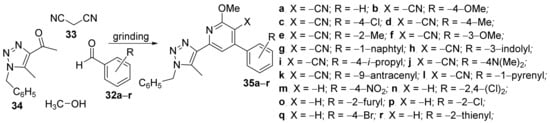
Jaabil et al.
[17] have studied the in vitro antimicrobial activities of some newly hybrid 1,2,3-triazolo-pyridine derivatives. The synthesis was green and efficient, under grinding strategy at room temperature, involving
one-pot sequential multicomponent reactions of aryl aldehydes
32a–
r, malonitrile
33, methanol and 1,2,3-triazolyl ketone
34, when the corresponding 1,2,3-triazolyl-pyridine/cyanopyridine hybrids
35a–
r were obtained,
Scheme 9.
Scheme 9. Reaction pathway to obtain 1,2,3-triazolo-pyridine hybrids 35a–r.
The synthesized 1,2,3-triazolo-pyridine hybrids
35a–
r were screened for their in vitro antibacterial activity against three human bacterial strains,
Staphylococcus aureus,
Salmonella typhi and
Escherichia coli, using the MIC method. Some of the 1,2,3-triazolyl cyanopyridine hybrids displayed a remarkable activity against the tested germs, better than tetracycline (standard drug), according to the R-substituent from the phenyl ring. The most active compounds were
35c (with R = −4-chloro-; MIC in the range of 50–90 μg/mL),
35e (with R = −2-methyl-; MIC in the range of 40–90 μg/mL) and
35r (with R = −2-thienyl; MIC in the range of 70–120 μg/mL). The hybrid 1,2,3-triazolo-pyridine compounds were also tested for their antioxidant activity in the assay by 2,2-diphenyl-1-picrylhydrazyl (DPPH) method, showing promising results.
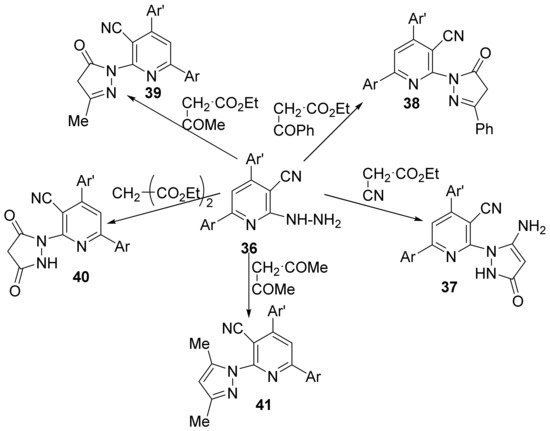
Felefel et al.
[18] synthesized three new series of some pyridine hybrid derivatives (namely pyrazole-pyridine
37–
41, triazolo-pyridine
42–
45 and triazino-pyridine
46) and studied their antimicrobial activities. The synthesis is using as starting material 6-(3,4-dimethylphenyl)-2-hydrazinyl-4-(thiophen-2-yl)-pyridine-3-carbonitrile
36 which react with different compounds with methylene active group (namely acetyl acetone, diethylmalonate, ethyl cyanoacetate, ethyl benzoylacetate and/or ethyl acetoacetate) to produce the desired pyrazole-pyridine hybrid derivatives
37–
41,
Scheme 10.
Scheme 10. Reaction pathway to obtain pyrazole-pyridine hybrids 37–41.
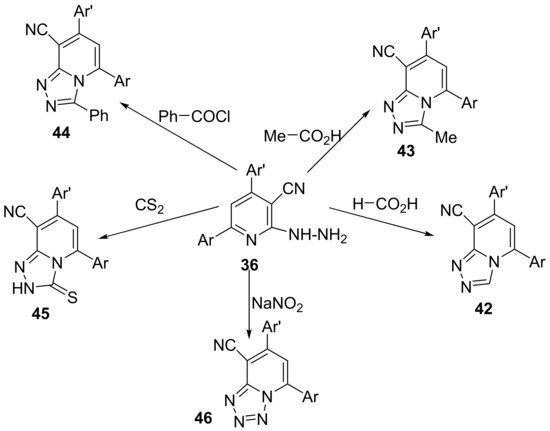
The synthesis of triazolo-pyridines
42–
45 and tetrazolo-pyridines
46 use as starting material the same intermediate, the 6-(3,4-dimethylphenyl)-2-hydrazinyl-4-(thiophen-2-yl)-pyridine-3-carbonitrile
36, which react with the appropriate formic acid, acetic acid, benzoyl chloride, carbon disulfide, respectively, sodium nitrite, to produce the desired hybrid derivatives
42–
45 and
46,
Scheme 11.
Scheme 11. Reaction pathway to obtain triazolo-pyridines 42–45 and tetrazolo-pyridine 46 hybrids.
The synthesized pyridine hybrids
37–
46 were screened for their in vitro antibacterial activity against
Gram-positive bacteria (
Staphylococcus aureus and
Bacillus subtilis),
Gram-negative bacteria (
Salmonella typhi and
Escherichia coli) and fungus (
Aspergillus flavus and
Candida albicans) using the disk diffusion agar technique. Some of the hybrids have significant antimicrobial activity, the most active compounds being
37 with a DIZ in the range of 10–17 mm. The antioxidant activity was also tested.
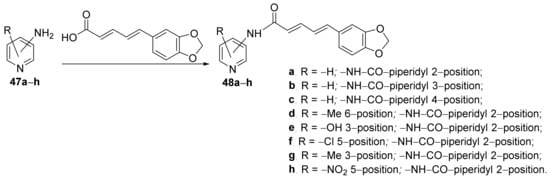
Amperayani et al.
[19] synthesized a library of piperine-pyridine hybrid derivatives and studied their antimicrobial activities. The reaction pathway is straight, in one step, involving an acylation reaction of various amino-pyridine derivatives
47a–
h, when the corresponding hybrids piperine-pyridine derivatives
48a–
h are obtained,
Scheme 12.
Scheme 12. Reaction pathway to obtain piperine-pyridine hybrids 48a–h.
The synthesized piperine-pyridine hybrid derivatives
48a–
h were tested for their in vitro antibacterial activity against some
Gram-positive and
Gram-negative bacterial strains (
Bacillus subtilis,
Streptobacillus,
Staphylococcus aureus,
Escherichia coli,
Klebsiella pneumoniae,
Pseudomonas aeruginosa,
Enterococcus faecalis and
Salmonella typhi) and fungus strains (
Aspergillus niger,
Aspergillus flavus,
Aspergillus fumigatus and
Candida albicans) using the disk diffusion agar technique. The piperine-pyridine hybrids
48a,
48d and
48h have very good activity against the
Gram-negative strains
E. coli,
K. pneumoniae,
E. faecalis and
P. aeruginosa, having a DIZ in the range of 22–26 mm, superior to control standard drug). The antifungal activity of hybrids was moderate.
3. Six-Member Ring Azaheterocycles with One Nitrogen Atom. Hybrid Quinoline and Isoquinoline
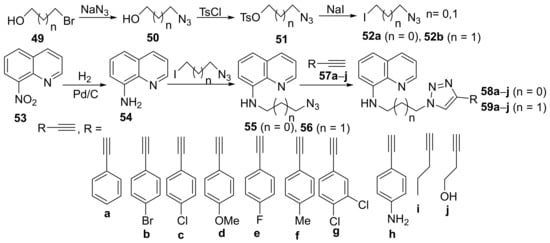
In their attempt to obtain new quinoline derivatives with antimicrobial activity, Albayrak et al.
[20] synthesized a library of 20 new triazolo-quinoline hybrid derivatives and studied their antimicrobial activities. The reaction pathway involves several steps (
Scheme 13), starting from 8-nitroquinoline
53. The initial reduction reaction of
53 is leading to 8-aminoquinoline
54, which is suffering a subsequent
N-alkylation with azido-iodo-propane
52a,
b (generated from the corresponding bromo-alkyl alcohol) leading to alkyl-azide-quinolines
55 and
56. Finally, the alkyl-azide-quinoline derivatives are treated with the corresponding alkyne
57a–
j leading to the desired products, the triazolo-quinoline hybrid derivatives
58a–
j and
59a–
j.
Scheme 13. Reaction pathway to obtain triazolo-quinoline hybrids 58a–j and 59a–j.
The synthesized triazolo-quinoline hybrid derivatives
58a–
j and
59a–
j were tested for their in vitro antibacterial activity against some
Gram-positive and
Gram-negative bacterial strains (
Bacillus subtilis,
Streptococcus pneumoniae,
Staphylococcus aureus,
Escherichia coli,
Klebsiella pneumoniae,
Pseudomonas aeruginosa and
Enterococcus faecalis) and fungus strains (
Candida parapsilosis and
Candida albicans) using the disk diffusion agar technique. The triazolo-quinoline hybrid derivatives
58a–
j and
59a–
j manifest good activity against the tested strains. The most active compound was
58a, having excellent activity against
E. coli,
P. aeruginosa,
K. pneumoniae,
E. faecalis,
S. aureus,
S. pneumoniae,
B. subtilis,
C. albicans and
C. parapsilosis. In some cases, the activity was several orders of magnitude superior to control drugs (DIZ of
58a was in the range of 35–250 mm; control, ampicillin, respectively, fluconazole have had a DIZ of 35 mm).
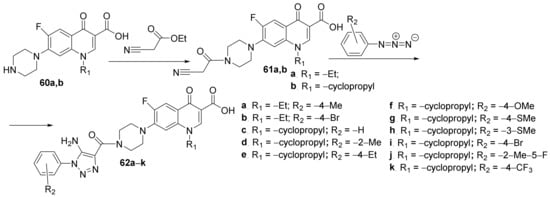
Hryhoriv et al.
[21][22] synthesized two new classes of hybrid derivatives analogous to fluoroquinolones, namely piperidino-quinoline
61a,
b and 1,2,3-triazolo-piperidino- quinoline
62a–
k, and studied their antimicrobial activities. The first class of hybrids was obtainedviaan
N-alkylation reaction of piperidino-quinoline
60a,
b, when the
N-substituted-piperidino-quinoline hybrids
61a,
b are obtained. A click cyclocondensation reaction of
61a,
b occurs to the second class of hybrids, the 1,2,3-triazolo-piperidino-quinoline
62a–
k,
Scheme 14.
Scheme 14. Reaction pathway to obtain N-substituted-piperidino-quinoline hybrids 61a,b and1,2,3-triazolo-piperidino-quinoline hybrids 62a–k.
The synthesized hybrid derivatives piperidino-quinoline
61a,
b and 1,2,3-triazolo-piperidino-quinoline
62a–
k were tested for their in vitro antibacterial activity against standard bacterial strains
Staphylococcus aureus and
Escherichia coli, respectively, and the fungus
Candida albicans using the disk diffusion agar technique. The antimicrobial assay was also made by some clinical bacterial strains
S. aureus and
E. coli, respectively, and fungus
C. albicans using the same method. The hybrid, 1,2,3-triazolo-piperidino-quinoline
62c have a very good activity against the tested standard strains (DIZ in the range of 25–35 mm), having a superior inhibition zone to control (DIZ = 25 mm). Against clinical microbial strains, the activity was negligible.
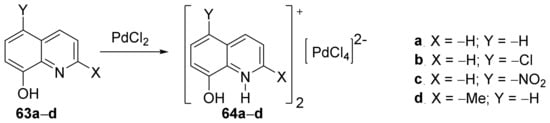
Drweesh et al.
[23] synthesized hybrid organic-inorganic derivatives and studied their antimicrobial activities, antiproliferative activity, and radical scavenging properties. In order to synthesize the desired palladium-quinoline derivatives
64a–
d, they used organic cation modulation, doing a complexation reaction with PdCl
2 of the quinolines
63a–
d,
Scheme 15.
Scheme 15. Reaction pathway to obtain metal-quinoline hybrids 64a–d.
The synthesized palladium-quinoline derivatives hybrids
64a–
d and the free ligands
63a–
d, were tested for their in vitro antimicrobial activity against 14 standard microbial strains (
Gram-positive and
Gram-negative bacteria, fungus:
Bifidobacterium animalis,
Lactobacillus plantarum,
Bacillus subtilis,
Staphylococcus aureus ATCC 663,
Staphylococcus aureus ATCC 25923,
Pseudomonas aeruginosa,
Proteus mirabilis ,
Escherichia coli,
Salmonella enterica,
Candida albicans,
Saccharomyces boulardii,
Aspergillus flavus,
Trichoderma viridae,
Aspergillus niger). All hybrid compounds
64a–
d have high antimicrobial activity against all tested strains, with minimum inhibitory concentration values ranging from 1.95 to 250 μg/mL. The results of DNA interaction studies indicate that the hybrids
64a–
d and the free ligands
63a–
d, interact with the DNAvia an intercalation mechanism (the aromatic chromophore intercalates the base pairs of DNA; compound
64a has the highest binding affinity). The anticancer activity was also studied, with compounds
64a and
64b having selective and high cytotoxicity against human lung and breast cancer cells.
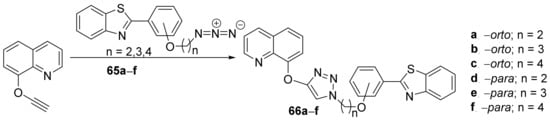
Nehra et al.
[24] synthesized a series of triazole-benzothiazole-quinoline hybrids and studied their antimicrobial properties. The reaction pathway is straight and efficient (
Scheme 16), involving a click cyclocondensation reaction of azido-alkyl-benzothiazole
65a–
f (generated in situ from the corresponding bromo-alkyl derivative) with the corresponding alkyne-quinoline, leading to the desired products, triazole- -benzothiazole-quinoline hybrids
66a–
f.
Scheme 16. Reaction pathway to obtain triazole-benzothiazole-quinoline hybrids 66a–f.
The synthesized hybrids
66a–
f were evaluated for their in vitro antimicrobial activity against two
Gram-positive strains (
Staphylococcus aureus and
Bacillus subtilis) and two
Gram-negative strains (
Escherichia coli and
Pseudomonas aeruginosa) and two fungal strains (
Candida tropicalis and
Aspergillus terreus). The tested hybrids have good antimicrobial activity against both bacteria and fungus. The most promising compound was proved to be
66a, with an antibacterial (DIZ in the range of 15–17 mm) and antifungal (DIZ in the range of 21–34 mm) activity superior to reference ciprofloxacin (DIZ = 22 mm) and fluconazole (DIZ = 20 mm), respectively. Interesting molecular docking studies were also performed.
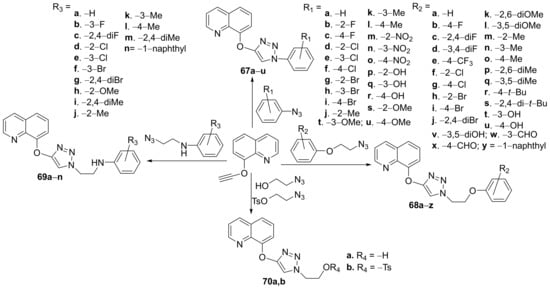
Awolade et al.
[25] synthesized a library of triazole-quinoline hybrids and studied their antimicrobial properties. The reaction pathway is straight involving click chemistry of various azides with triple bond derivatives,
via copper(I)-catalyzed azide-alkyne 3 + 2 dipolar cycloaddition reactions,
Scheme 17.
Scheme 17. Reaction pathway to obtaintriazole-quinoline hybrids 67a–u, 68a–z, 69a–n and 70a,b.
The synthesized hybrids
67a–
u,
68a–
z,
69a–
n and
70a,
b were evaluated for their in vitro antimicrobial activity against ESKAPE microbial strains (bacteria and fungus: (
Staphylococcus aureus,
Escherichia coli,
Acinetbacter baumannii,
Klebsiella pneumoniae,
Candida albicans and
Candida neoformans). Some of the compounds proved to have a good and broad-spectrum of antibacterial activity, against methicillin-resistant
S. aureus (MRSA),
E. coli,
A. baumannii, multidrug-resistant
K. pneumoniae and the fungus
C. albicans and
C. neoformans (superior to control, fluconazole). The most promising antibacterial compound was proved to be
70b with an MIC = 75.39 μM against MRSA,
E. coli,
A. baumannii, and multidrug-resistant
K. pneumoniae. The hybrid
70b also has a very good antifungal activity against
C. albicans and
C. neoformans with an MIC of 37.69 and 2.36 μM, respectively, superior to control fluconazole.
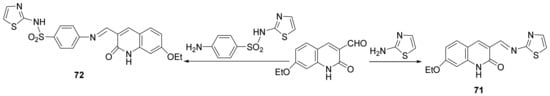
Ammar et al.
[26] synthesized a series of thiazole-quinoline hybrids and studied their antimicrobial properties. In order to synthesize the desired compounds, they used the condensation reaction between formil-quinoline derivatives with amino-thiazole or sulfathiazole, when the desired Schiff’s base thiazole-quinoline
71 and
72, are obtained,
Scheme 18.
Scheme 18. Reaction pathway to obtain thiazole-quinoline hybrids 71 and 72.
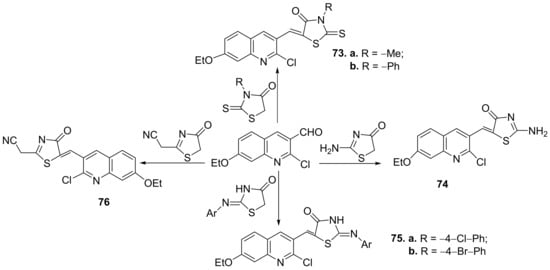
Further, the condensation reaction between formil-quinoline derivatives with different thiazolone derivatives lead to hybrid thiazolone-quinoline derivatives
73–
76,
Scheme 19.
Scheme 19. Reaction pathway to obtain thiazolone-quinoline hybrids 73–76.
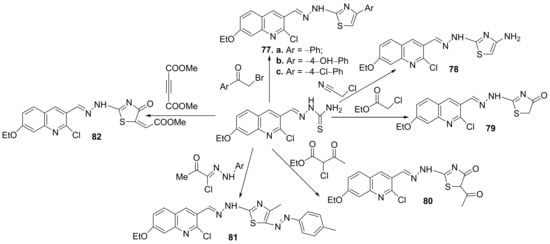
Finally, the cyclization of different quinoline-thiosemicarbazone derivatives with the halogenated compounds lead to other hybrid thiazole-quinoline derivatives
77–
82,
Scheme 20.
Scheme 20. Reaction pathway to obtain thiazole-quinoline hybrids 77–82.
The synthesized hybrids
71–
82, were evaluated for theirin vitroantimicrobial activity against eight standard microbial strains, three
Gram-positive bacteria (
Staphylococcus aureus,
Bacillus faecalis and
Bacillus subtilis), three
Gram-negative bacteria (
Escherichia coli,
Salmonella typhi and
Pseudomonas aeruginosa), and two fungi (
Candida albicans and
Fusarium oxysporum). Some of the compounds have good antimicrobial activity, with MIC and MBC values ranging between 0.95 and 62.5 µg/mL, and 1.94 and 118.7 µg/mL, respectively. Two compounds, namely
77b and
73a, proved to be the most active of the series against
S. aureus and
E. coli having an MIC between 0.95 and 7.81 μg/mL, respectively a MBC between 3.31 and 15.62 μg/mL.
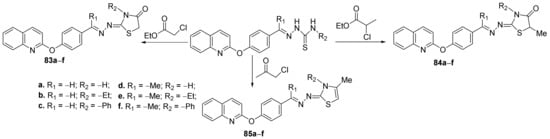
Using a similar strategy, some of the above researchers (Eissa et al.
[27]) synthesized a new series of thiazole-quinoline hybrids and studied their antimicrobial properties. In order to synthesize the desired compounds, they used the cyclization of quinoline-thiosemicarbazone derivatives with the halogenated compounds, when the corresponding hybrid thiazole-quinoline derivatives,
83a–
f,
84a–
f and
85a–
f are obtained,
Scheme 21.
Scheme 21. Reaction pathway to obtain thiazole-quinoline hybrids 83–85a–f.
The synthesized hybrids
83a–
f,
84a–
f and
85a–
f, were evaluated for their in vitro antimicrobial activity against
Gram-positive (five strains:
Staphylococcus aureus,
Staphylococcus epidermidis,
Streptococcus pyogenes,
Bacillus subtilis and
Enterococcus faecalis) and
Gram-negative bacteria (five strains:
Neisseria gonorrhoeae,
Proteus vulgaris,
Klebsiella pneumonia,
Shigella flexneri and
Pseudomonas aeruginosa), as well as fungus (five strains:
Aspergillus fumigatus,
Aspergillus clavatus,
Candida albicans,
Geotrichum candidum, and
Penicillium marneffei). Some of the compounds displayed good antimicrobial activity, superior to the used control. The most active compound was found to be
85e, having a two-fold potency compared with gentamycin for inhibition of
N. gonorrhoeae, four-fold potency compared with amphotericin B for the inhibition of
A. fumigatus, equipotent activity compared with the reference drugs for inhibition of
S. flexneri,
S. pyogenes,
P. vulgaris,
A. clavatus,
G. candidum and
P. marneffei.
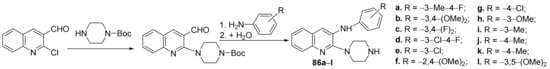
Lagdhir et al.
[28] synthesized a library of piperazin-quinoline hybrids and studied their antimicrobial properties. The reaction pathway involves two steps (an alkylation and a condensation reaction), leading to the piperazin-quinoline hybrids
86a–
l,
Scheme 22.
Scheme 22. Reaction pathway to obtain piperazin-quinoline hybrids 86a–l.
The synthesized hybrids
86a–
l were evaluated for their in vitro antibacterial (
Staphylococcus aureus,
Streptococcus pyogenes,
Escherichia coli and
Pseudomonas aeruginosa) and antifungal (
Aspergillus clavatus,
Aspergillus niger and
Candida albicans) activity, antimalarial (
Plasmodium falciparum) and antituberculosis (
Mycobacterium tuberculosis) activity. Some of the compounds have good antibacterial and antifungal activity against
S. aureus and
C. albicans. The hybrids
86a,
86b,
86d,
86j and
86k, are the most active as an antimicrobial against
S. aureus, having an MIC = 100 μg/mL, equal to the control drug ampicillin. The hybrid
86k has excellent antifungal activity against
C. albicans, having an MIC = 250 μg/mL, two folds higher compared with the control drug griseofulvin. The antimalarial and antitubercular activity proved to be moderate for the majority of compounds.
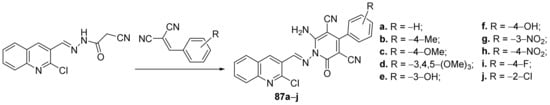
Desai et al.
[29] synthesized a series of pyridine-quinoline hybrids and evaluated it for their antimicrobial properties. The reaction pathway involves a cyclocondensation reaction of quinoline derivative with benzylidene-malononitril, when the corresponding pyridine-quinoline hybrids
87a–
j were obtained,
Scheme 23.
Scheme 23. Reaction pathway to obtain pyridine-quinoline hybrids 87a–j.
The synthesized hybrids
87a–
j were evaluated for their in vitro antimicrobial activity against
Gram-positive (two strains:
Staphylococcus aureus and
Staphylococcus pyogenes) and
Gram-negative (two strains:
Escherichia coli and
Pseudomonas aeruginosa) bacteria, as well as to fungus (three strains:
Aspergillus clavatus,
Aspergillus niger and
Candida albicans). Some of the compounds displayed promising antimicrobial activity. The hybrid
87i has the best antibacterial activity against
E. coli,
P. aeruginosa and
S. aureus strains, with an MIC = 12.5 μg/mL, two folds higher compared with the control drug ciprofloxacin (MIC = 25 μg/mL). The most active compound against
C. albicans was found to be
87e, having an MIC=25 μg/mL, much better compared with the control drug griseofulvin (MIC = 500 μg/mL).
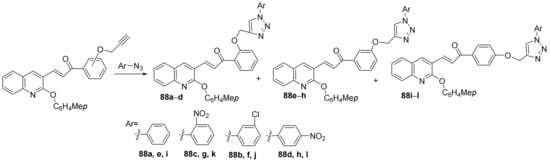
Vishnuvardhan et al.
[30] synthesized a library of triazole-quinoline hybrids and studied their antimicrobial properties. The reaction pathway involves a typical click cyclocondensation reaction of quinoline with a triple bond with aryl-azide derivatives, when the corresponding triazole-quinoline hybrids
88a–
l,
Scheme 24.
Scheme 24. Reaction pathway to obtain triazole-quinoline hybrids 88a–l.
The synthesized hybrids
88a–
l were evaluated for theirin vitroantimicrobial activity against
Gram-positive (
Staphylococcus aureus and
Enterococcus faecalis) and
Gram-negative (
Escherichia coli and
Pseudomonas aeruginosa) bacteria, as well as to fungus (
Aspergillus niger and
Candida albicans). Most of the hybrid compounds have good antimicrobial activity. The best antibacterial activity reveals the hybrids
88d,
88h and
88i, having a DIZ in the range of 16–21 mm, superior to control ampicillin (DIZ = 15 mm). The best antifungal activity reveals the hybrids
88d,
88h and
88k, having a DIZ in the range of 18–27 mm, superior to control griseofulvin (DIZ = 17 mm).
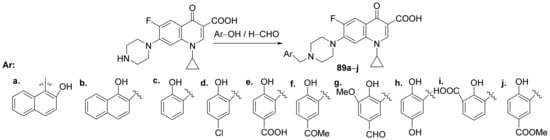
Abdel-Rahman et al.
[31] synthesized a series of piperazin-quinoline hybrids derived from ciprofloxacin and studied their antimicrobial and anticancer properties. The reaction pathway involves the reaction of ciprofloxacin with the corresponding phenolic derivatives with an excess of formaldehyde, when the piperazin-quinoline hybrids
89a–
j are obtained,
Scheme 25.
Scheme 25. Reaction pathway to obtain piperazin-quinoline hybrids 89a–j.
The synthesized hybrids
89a–
j were evaluated for their antimicrobial and anticancer activity. The antibacterial screening was preconformed on
Gram-positive and
Gram-negative strains:
Staphylococcus aureus, MRSA clinical strain, MRSA reference strain,
Escherichia coli and
Pseudomonas aeruginosa. The obtained results reveal that the hybrid
89d has the best antibacterial activity against
S. aureus, MRSA (reference strain) and MRSA (clinical strain) with an MIC of 0.57, 0.52, and 0.082 µg/mL, respectively, (compared with the reference standard drug ciprofloxacin which has an MIC of 1.63 µg/mL against
S. aureus, an MIC of 1.45 µg/mL against MRSA reference, and an MIC of 0.84 µg/mL against MRSA clinical). The hybrid
89j exhibited the best antimicrobial activity against
E. coli and
P. aeruginosa, with an MIC of 0.036 and 0.043, respectively, (compared with the reference standard drug ciprofloxacin which has an MIC of 0.056 µg/mL against
E. coli and an MIC of 1.27 µg/mL against
P. aeruginosa).
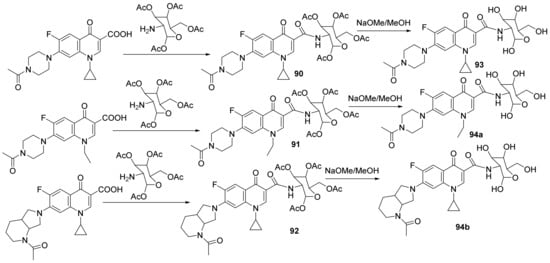
Mohammed et al.
[32] synthesized a series of glycosylated-quinoline hybrids derived from fluoroquinolone and studied their antimicrobial properties. The reaction pathway involves the reaction of ciprofloxacin with the corresponding phenolic derivative with an excess of formaldehyde, when the glycosylated-quinoline hybrids
90–
94 are obtained,
Scheme 26.
Scheme 26. Reaction pathway to obtainglycosylated-quinoline hybrids 90–94.
The synthesized glycosylated-quinoline hybrids
90–
94 were evaluated for their antibacterial activity against various
Gram-positive and
Gram-negative bacteria:
Escherichia coli,
Listeria monocytogenes,
Salmonella enterica,
Pseudomonas aeruginosa,
Listeria monocytogenes,
E. coli clinical isolate (resistant to nalidixic acid, ciprofloxacin HCl and norfloxacin antibiotics), methicillin-resistant
Staphylococcus aureus (MRSA), methicillin-sensitive
Staphylococcus aureus (MSSA). The hybrids were also tested for their antifungal activity against fungi:
Candida albicans,
Aspergillus flavus,
Fusarium solani,
Stachybotrys chartarum and
Penicillium chrysogenum. The hybrid compounds
90,
91 and
94a have excellent antimicrobial activity against a fluoroquinolone-resistant
E. coli clinical isolate, comparable to controls ciprofloxacin and norfloxacin. The hybrid compound
91 also has good antifungal activity against
C. albicans and
P. chrysogenum.
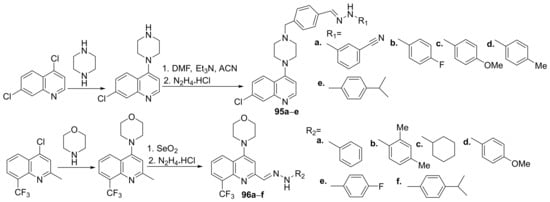
Shruthi et al.
[33] synthesized a series of piperazine-quinoline hybrids
95a–
e and morpholine-quinoline hybrids
96a–
f and evaluate them for their antimicrobial properties. The reaction pathway is depicted in
Scheme 27.
Scheme 27. Reaction pathway to obtain piperazine- and morpholine-quinoline hybrids 95a–e and 96a–f.
The synthesized hybrids
95a–
e and
96a–
f were evaluated for their antibacterial (
Acinetobacter baumanii,
Enterococcus faecium,
Klebsiella pneumonia,
Pseudomonas aeruginosa,
Escherichia coli and
Staphylococcus aureus) and antitubercular (
Mycobacterium tuberculosis) activity. Hybrid
95b has the best antibacterial activity against
E. coli and
S. aureus strains with an MIC of 4, respectively, 2 µg/mL, compared to standard drug vancomycin (MIC of 16, respectively, 0.5 µg/mL). Hybrids
95d,
95e and
96f exhibited the best antibacterial activity against
A. baumaniistains with an MIC in the range of 1–2 µg/mL, compared to standard drug vancomycin (MIC = 0.5 µg/mL). Hybrids
95b,
95d and
95e also have promising antitubercular activity with an MIC of 4 µg/mL.
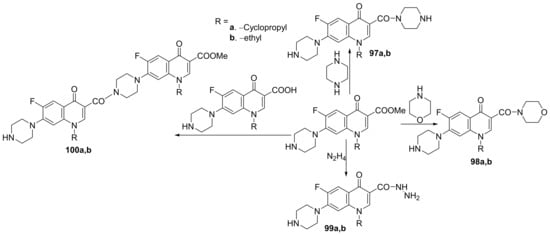
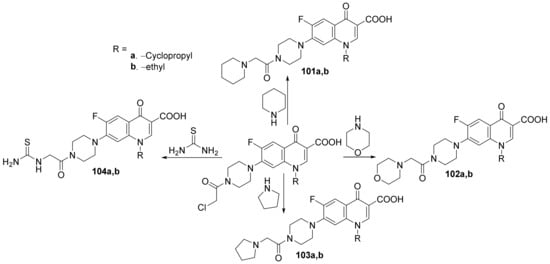
Kaur et al.
[34] synthesized a series of 3- and 7- substituted-quinoline hybrids derived from fluoroquinolone and studied their antimicrobial properties. The reaction pathway involves the reaction of fluoroquinolone derivatives with the corresponding reagents, when the quinoline hybrids
97–
104a,
b are obtained,
Scheme 28 and
Scheme 29.
Scheme 28. Reaction pathway to obtain piperazino-quinoline hybrids 97–100a,b.
Scheme 29. Reaction pathway to obtain 7-substituted-quinoline hybrids 101–104a,b.
The synthesized quinoline hybrids
97–
104a,
b were evaluated for their antibacterial activity against four bacterial strains:
Bacillus subtilis,
Pseudomonas aeruginosa,
Escherichia coli and
Staphylococcus aureus. All hybrids
97–
104a,
b have proved to be active against all bacterial strains, with an MIC value of 25 μg/mL which is fourfold more active compared to the standard drug ciprofloxacin (MIC = 100 μg/mL).
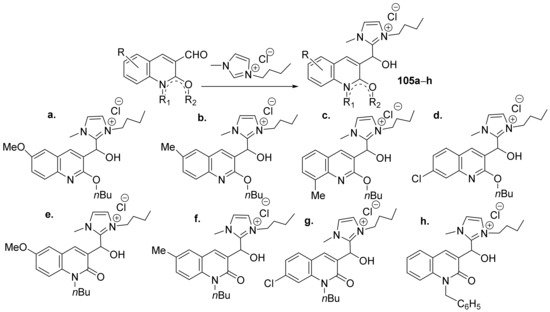
Insuasty et al.
[35] synthesized a series of imidazolium-quinoline hybrids and studied their antimicrobial properties. The reaction pathway involves the reaction of 3-formyl-quinolone derivatives with the corresponding imidazolium salts, when the imidazolium-quinoline hybrids
105a–
h are obtained,
Scheme 30.
Scheme 30. Reaction pathway to obtain imidazolium-quinoline hybrids 105a–h.
The synthesized imidazolium-quinoline hybrids
105a–
h were evaluated for their antibacterial (
Klebsiella pneumoniae,
Escherichia coli and
Staphylococcus aureus), antifungal (
Cryptococcus neoformans) and antitubercular (
Mycobacterium tuberculosis H37Rv and
Mycobacterium bovis BCG) activities. Hybrid derivatives
105c,d demonstrated a remarkable antifungal activity against
C. neoformans (MIC in the range of 15 µg/mL) while for the other fungal strains the activity is weak. The hybrids have modest antibacterial activity (both against
Gram-positive and
Gram-negative bacteria) as well as antitubercular activity.
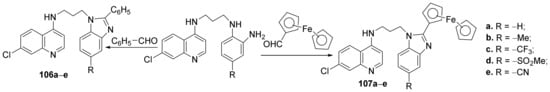
Baartzes et al.
[36] synthesized a series of benzimidazole-quinoline and ferrocenyl-quinoline hybrids and studied their antimicrobial properties. The reaction pathway involves the reaction of amino-quinoline derivatives with the corresponding formyl derivatives, when the benzimidazole-quinoline hybrids
106a–
e and ferrocenyl-quinoline hybrids
107a–
e are obtained,
Scheme 31.
Scheme 31. Reaction pathway to obtain benzimidazole-quinoline and ferrocenyl-quinoline hybrids 106a–e and 107a–e.
The synthesized quinoline hybrids
106a–
e and
107a–
e were evaluated for their antimalarial (
Plasmodium falciparum and
Plasmodium berghei) and antitubercular (
Mycobacterium tuberculosis) activity. All hybrid derivatives are active against tested malaria strains and have modest activity against them. The most active hybrids against malarial strains have proved to be
106c and
107b, with an IC
50 of 0.43, respectively, 0.32 µM, compared with the standard drug chlorquine (IC
50 = 0.01 µM).
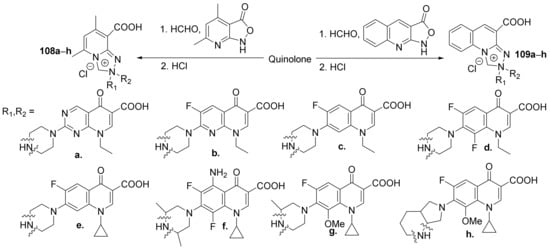
Fedorowicz et al.
[37] synthesized a series of zwiterionic hybrids pyridine-fluoroquinolone
108a–
h and quinoline-fluoroquinolone
109a–
h and studied their antimicrobial properties. The reaction pathway involves a tandem Mannich-electrophilic amination reaction of isoxazolones derivatives and fluoroquinolone bearing a secondary amino group at position 7 of the quinoline ring,
Scheme 32.
Scheme 32. Reaction pathway to obtain zwiterionic pyridine-fluoroquinolone and quinoline-fluoroquinolone hybrids 108a–h and 109a–h.
The synthesized quinoline hybrids
108a–
h and
109a–
h were evaluated for their antibacterial activity against
Gram-positive and
Gram-negative bacterial strains (laboratory and clinical:
Staphylococcus aureus ATCC 6538,
Staphylococcus aureus MRSA N315,
Staphylococcus epidermidis ATCC 14990,
Bacillus subtilis ATCC 6633,
Escherichia coli ATCC 8739,
Pseudomonas aeruginosa ATCC 9027,
Proteus vulgaris NCTC 4635,
Staphylococcus aureus MRSA 6347,
Staphylococcus epidermidis MRSE 13199 and
Serratia marcescens 12795) as well as for antibiofilm activity. The hybrid derivatives proved to have bactericidal and antibiofilm activity. The most active hybrids were found to be
109d and
109e, exhibiting good inhibition against all strains, with the IC
50 values in the low micromolar range.
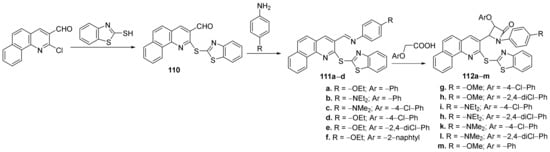
Borazjani et al.
[38] synthesized a library of quinoline hybrids (benzothiazole-benzo-quinoline
110, imino-benzothiazole-benzo-quinoline
111a–
d, β-lactam-benzo-thiazole-benzo-quinoline
112a–
m) and studied their antimicrobial properties. The reaction pathway involves a [2+2]-cycloaddition reaction of imines
111a–
d and ketenes derived from substituted acetic acids,
Scheme 33.
Scheme 33. Reaction pathway to obtain benzothiazole-benzo-quinoline hybrids 110, 111a–d and 112a–m.
The synthesized quinoline hybrids
110–
112 were evaluated for their antimicrobial activity against
Gram-positive and
Gram-negative bacterial strains:
Staphylococcus aureus,
Bacillus subtilis,
Enterococcus faecalis,
Salmonella typhi,
Escherichia coli and
Pseudomonas aeruginosa. From the β-lactam class, the assay indicates that the most active hybrids against
E. coli and
P. aeruginosa, are
112k and
112m, with an MIC of 42, respectively, 20 μg/mL, compared to standard drug gentamycin (MIC of 90, respectively, 5 μg/mL). From the imino-benzothiazole-benzo-quinoline class, the most active hybrids against
P. aeruginosa and
S. aureus, are
111a–
c, with an MIC of 42 μg/mL, compared to standard drug gentamycin (MIC of 5, respectively, 90 μg/mL).
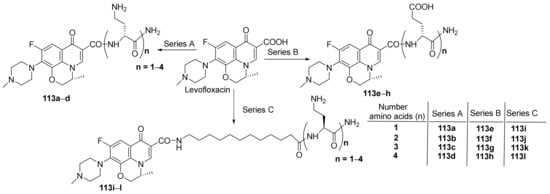
Berry et al.
[39] synthesized a series of peptide-fluoroquinolone hybrids and studied their antimicrobial properties. In order to synthesize the desired hybrids, the researchers used solid-phase peptide synthesis, from levofloxacin fluoroquinolone with the corresponding peptide (oligopeptide), when the desired peptide-fluoroquinolone hybrids
113a–
l are obtained,
Scheme 34.
Scheme 34. Reaction pathway to obtain peptide-quinolone hybrids 113a–l.
The synthesized peptide-fluoroquinolone hybrids
113a–
l were evaluated for their antimicrobial activity against MDR bacterial strains,
Gram-negative and
Gram-positive:
Pseudomonas aeruginosa,
Escherichia coli,
Klebsiella pneumoniae,
Acinetobacter baumannii, methicillin-resistant
Staphylococcus aureus (MRSA), methicillin-sensitive
Staphylococcus aureus (MSSA), methicillin-resistant
Staphylococcus epidermis (MRSE),
Enterococcus faecalis,
Enterobacter cloacae,
Stenotrophomonas maltophilia. The assay indicates that all the peptide-hybrids have weak antibacterial activity. If the hybrids are mixed with fluoroquinolone (ciprofloxacin, levofloxacin and moxifloxacin) drugs, the resulting conjugates possess antimicrobial activity against MDR
Gram-negative bacteria (clinical isolates,
P. aeruginosa,
E. coli,
K. pneumoniae,
A. baumannii), superior to reference levofloxacin.
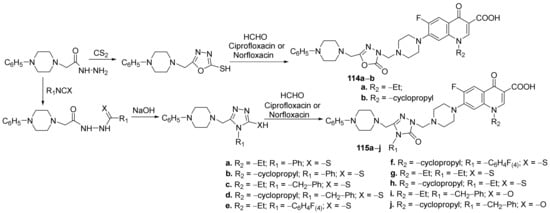
Mermer et al.
[40] synthesized a library of triazole- and oxadiazole-fluoroquinolone hybrids and studied their antimicrobial properties. The reaction pathway took placeviaseveral steps of sequential reactions, starting from phenyl piperazine. Finally, the corresponding triazole-fluoroquinolone
114a,
b and oxadiazole-fluoroquinolone
115a–
j hybrids were obtained
via a
one-pot three-component Mannich reaction,
Scheme 35. The reactions were performed both under conventional thermal heating and microwave, the last pathway being more advantageous.
Scheme 35. Reaction pathway to obtain oxadiazole- and triazole-fluoroquinolone hybrids 114a,b and 115a–j.
The synthesized hybrids
114a,
b and
115a–
j were tested for their antimicrobial activity (against
Gram-positive and
Gram-negative strains:
Staphylococcus aureus,
Enterococcus faecalis,
Escherichia coli,
Pseudomonas aeruginosa,
Klebsiella pneumoniae,
Acinetobacter haemolyticus), DNA gyrase and Topoisomerase IV inhibition potentials. The hybrids have good antimicrobial activity and displayed excellent DNA gyrase inhibition. The hybrids
114b,
115b and
115h exhibited the best antimicrobial activity against the tested strains. Thus, the hybrids have excellent activity against
K. pneumoniae with an MIC of 0.25 µg/mL, compared with the standard drug gentamycin (MIC = 0.25 µg/mL). The hybrids also have excellent activity against
A. haemolyticus and
P. aeruginosa with an MIC in the range of 0.5–2 µg/mL, compared with the standard drug gentamycin (MIC = 0.78 µg/mL, respectively, MIC = 1.56 µg/mL). Against
Gram-positive strain
E. faecalis the hybrids have excellent activity with an MIC in the range of 0.5–8 µg/mL, compared with the standard drug ampicillin (MIC = 12.5 µg/mL).
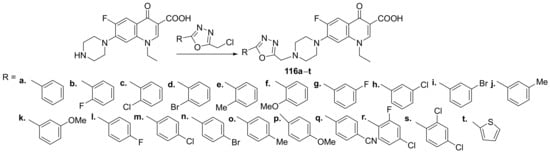
Guo et al.
[41] synthesized a library of oxadiazole-quinoline hybrids and studied their antibacterial properties. The reaction pathway is straight, involving an alkylation reaction of fluoroquinolone with the corresponding oxadiazole, when the desired oxadiazole-fluoroquinolone hybrids
116a–
t were obtained,
Scheme 36.
Scheme 36. Reaction pathway to obtain oxadiazole-fluoroquinolone hybrids 116a–t.
The synthesized oxadiazole-fluoroquinolone hybrids
116a–
t were tested for their antibacterial activity against methicillin-resistant
Staphylococcus aureus (MRSA) and laboratory
Staphylococcus aureus. The hybrids displayed good antibacterial activity, one of the compounds
116k exhibited excellent antibacterial activity against both methicillin-resistant
S. aureus and laboratory
S. aureus, with an MIC in the range of 0.25–2 μg/mL, superior to control drug vancomycin (MIC = 2 μg/mL).
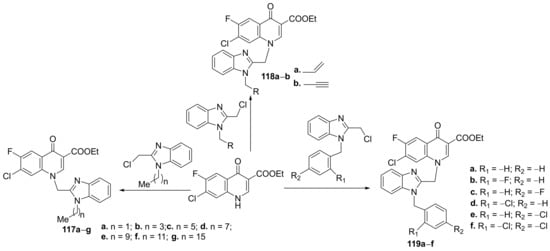
Wang et al.
[42] synthesized a series of benzimidazole–quinoline hybrids and studied their antibacterial and antifungal properties. The reaction pathway involves an
N-alkylation reaction of fluoroquinolone with the corresponding benzimidazole, when the desired benzimidazole-fluoroquinolone hybrids
117a–
g,
118a,
b and
119a–
f, were obtained,
Scheme 37.
Scheme 37. Reaction pathway to obtain benzimidazole-quinoline hybrids 117a–g, 118a,b and 119a–f.
The synthesized benzimidazole-fluoroquinolone hybrids
117a–
g,
118a,
b and
119a–
f were screened against
Gram-positive and
Gram-negative bacteria, respectively, fungus (methicillin-resistant
Staphylococcus aureus (MRSA),
Enterococcus faecalis,
Staphylococcus aureus,
Staphylococcus aureus ATCC25923,
Staphylococcus aureus ATCC29213,
Klebsiella pneumonia,
Escherichia coli,
Pseudomonas aeruginosa,
Acinetobacter baumanii,
Pseudomonas aeruginosa ATCC27853,
Escherichia coli ATCC25922,
Candida albicans,
Candida tropicalis,
Aspergillus fumigatus,
Candida albicans ATCC90023,
Candida parapsilosis ATCC22019). The results of the assay were promising, with some hybrids having excellent antibacterial activity. The most active hybrids against
K. pneumonia are
117a and
117c, with an MIC of 8 μg/mL, compared to the standard drug norfloxacin (MIC > 512 μg/mL).The most active hybrids against
S. aureus are
119a and
119f, with an MIC of 4 μg/mL, compared to the standard drug norfloxacin (MIC = 64 μg/mL).
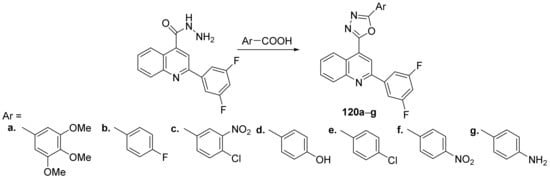
Bharadwaj et al.
[43] synthesized a series of oxadiazole–quinoline hybrids and studied their antibacterial and antifungal properties. The reaction pathway involves a cyclocondensation reaction of hydrazinyl-quinoline derivative with the corresponding aromatic acids, when the desired oxadiazole–quinoline hybrids
120a–
g were obtained,
Scheme 38.
Scheme 38. Reaction pathway to obtain oxadiazole-quinoline hybrids 120a–g.
The synthesized oxadiazole–quinoline hybrids
120a–
g were tested against clinical isolates
Gram-positive and
Gram-negative bacteria (
Staphylococcus aureus,
Bacillus cereus,
Escherichia coli,
Serratia marcescens), respectively, fungus (
Aspergillus niger,
Trichophyton mentagrophytes,
Candida albicans,
Candida parapsilosis). The antimicrobial activity of oxadiazole–quinoline derivatives was good, the hybrids
120a and
120f having the best antimicrobial activity against
B. cereus with an MIC of 17, respectively, 24 μg/mL, compared to standard drug ampicilin (MIC = 16 μg/mL).
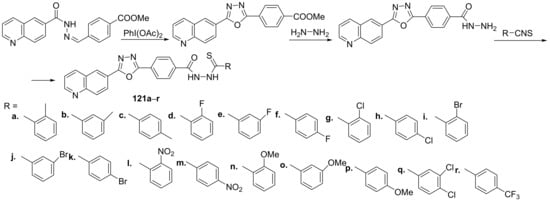
Tahaab et al.
[44] synthesized a series of oxadiazole–quinoline hybrids and studied their leishmanicidal potential. The reaction pathway to obtain the oxadiazole–quinoline hybrids
121a–
r is depicted in
Scheme 39.
Scheme 39. Reaction pathway to obtain oxadiazole-quinoline hybrids 121a–r.
The synthesized oxadiazole-quinoline hybrids
121a–
r were tested for their leishmanicidal activity against
Leishmania major promastigote. Most of the synthesized hybrids have a good leishmanicidal activity, compound
121r was found to be the most active (IC
50 = 0.10 μM) from the series, being 70 times more active than the standard drug (pentamidine, IC
50 = 7 μM).
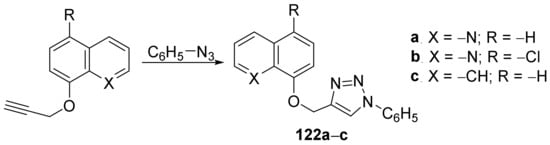
Irfan et al.
[45] synthesized a series of triazole–quinoline hybrids and studied their antifungal properties. The reaction pathway involves a typical click cyclocondensation reaction of azide with a compound with a triple bond, when the desired triazole–quinoline hybrids
122a–
c were obtained,
Scheme 40.
Scheme 40. Reaction pathway to obtain triazole-quinoline hybrids 122a–c.
The synthesized triazole–quinoline hybrids
122a–
c were tested against fungus
Candida albicans, both clinical isolates and laboratory strains [three FLC susceptible strains (
C. albicans D27,
C. albicans D31 and
C. albicans D39) and one FLC resistant strain (
C. albicans D15.9)]. The best antifungal activity was found for the hybrids
122a and
122b, having an MIC of 25 μg/mL for
122a and an MIC of 250 μg/mL for
122b, compared to control FLC (MIC >1 μg/mL)
Pandya et al.
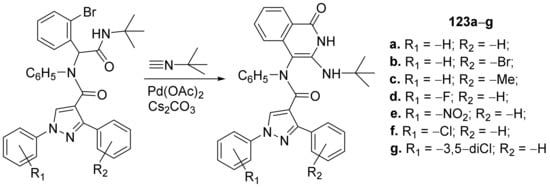
[46] synthesized a library of pyrazole–isoquinoline hybrids and studied their antimicrobial properties. The reaction pathway involves a palladium-catalyzed reaction of pyrazole derivatives with
t-butyl-isocyanide, when the corresponding pyrazole–isoquinoline hybrids
123a–
g, were obtained,
Scheme 41.
Scheme 41. Reaction pathway to obtain pyrazole-isoquinoline hybrids 123a–g.
The synthesized pyrazole–isoquinoline hybrids
123a–
g were evaluated for their antimicrobial activity against different pathogenic strains: bacterial strains (
Staphylococcus aureus,
Escherichia coli,
Enterococcus faecalis,
Streptococcus pyogens and
Vibrio cholera), fungal strains (
Candida albicans,
Candida glabrata,
Candida krusei,
Candida tropicalis and
Candida parapsilosis) and tubercular strain (
Mycobacterium tuberculosis). The antimicrobial activity of hybrids was very good, the hybrids
123e and
123g having the best antimicrobial activity, compared to standard drugs kanamycin and amphotericin B. Thus, the most active hybrids against
S. aureus are
123e and
123g, having an MIC of 20 μM, respectively, 37 μM, compared to standard drug kanamycin (MIC of 31 μM). The most active hybrids against
V. cholera are
123e and
123g, having an MIC of 41 μM, respectively, 90 μM, compared to the standard drug kanamycin (MIC of 62 μM). The hybrids
123e and
123g have the best antitubercular activity against
M. tuberculosis with an MIC of 30 μg/mL, respectively, 32 μg/mL, compared to standard drugs rifampicin and isoniazide (MIC of 90 μg/mL).
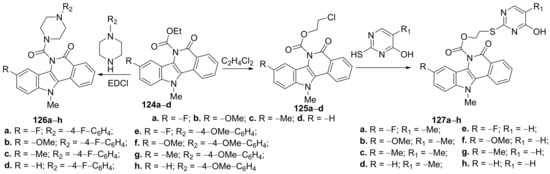
Verma et al.
[47] obtained a series of piperazine- and pyrimidine- isoquinoline hybrids and studied their antimicrobial properties. The piperazine-isoquinoline hybrids
126a–
h were synthesized by condensation of the carboxylic acid intermediates
124a–
d with appropriate aryl-piperazines,
Scheme 38. The pyrimidine-isoquinoline hybrids
127a–
h were synthesized in two steps: an
O-alkylation of the carboxylic acid intermediates
124a–
d (with ethylene dichloride), followed by an
S-alkylation of the obtained compounds
125a–
d (with thio-pyrimidine),
Scheme 42.
Scheme 42. Reaction pathway to obtain piperazine- and pyrimidine-isoquinoline hybrids 126a–h and 127a–h.
The synthesized piperazine- and pyrimidine-isoquinoline hybrids
124a–
h and
125a–
h were evaluated for their antibacterial and antifungal (
Escherichia coli,
Klebsiella pneumoniae,
Staphylococcus aureus,
Bacillus subtilis,
Aspergillus niger,
Aspergillus oryzae,
Candida albicans and
Pencillium chrysogenum), antioxidant, anticancer and antituberculosis (
Mycobacterium tuberculosis) activities. The antibacterial assay indicates that three hybrids, namely
124a,
125a and
126e have the best activity against
E. coli (with an MIC in the range of 1–3 μg/mL) and
K. pneumoniae (with an MIC in the range of 1.5–3 μg/mL), compared with the standard drug ciprofloxacin (MIC = 1.5 μg/mL). The hybrids
125a,
126a and
127a also have excellentactivity against
S. aureus (with an MIC in the range of 1–3 μg/mL) and
B. subtilis (with an MIC in the range of 1.5–3 μg/mL), compared with the standard drug ciprofloxacin (MIC = 1.5 μg/mL, respectively, MIC = 3 μg/mL). The hybrids
125a,
126a and
127a have excellent activity against fungus
A. niger,
C. albicans,
A. oryzae, and
P. chrysogenum (with an MIC of 1.5 μg/mL), compared with the standard drug fluconazole (MIC = 1.5 μg/mL for
A. niger and
C. albicans, respectively, MIC = 3 μg/mL for
A. oryzae, and
P. chrysogenum). The hybrids
127b and
127e have the best activity against
M. tuberculosis (MIC 1.0 mg/mL), compared with the standard drug rifampicin (MIC = 0.1mg/mL. The antioxidant and anticancer activity proved to be modest.