1. Introduction
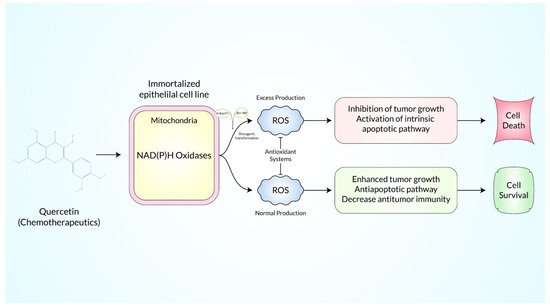
Due to its antioxidant properties, quercetin is used therapeutically for a variety of disorders. Mainly, the scavenging activity of
reactive oxygen species (ROS
) is converted to hydroxyl ions (OH
−) and holds the electron exchange capability
[1][136].
Quercetin (QC
) is lipophilic in nature and can easily pass through the cell membranes and latterly activate numerous intracellular signaling pathways, which are health effective
[2][137]. It has been demonstrated that it mediates not only intrinsic (mainly mitochondrial) but also extrinsic (Fas/FasL) apoptotic cancer cell death (
Figure 1)
[3][4][57,138]. In addition to apoptosis, previous research has suggested that QC plays an important role in arresting the cell cycle and controlling the expression of CDK enzyme (cyclin-dependent kinases)
[5][139]. Recent reports mention that it hinders the activity of cytochrome P450 (CYP) enzymes in hepatocytes and that most of them have crucial roles in the metabolism of drugs
[6][140]. QC has also been shown to suppress metastatic protein expression such as MMPs (matrix metalloproteases)
[7][50]. Actually, the metastasis process is also supported via neo-angiogenesis, and interestingly, QC is also noted to hinder neovascularization within the microenvironment of tumors
[8][141]. Another antitumor property of QC is its ability to inhibit inflammatory mediators including IFN-γ, IL-6, COX-2, IL-8, iNOS, TNF-α, and many other cancer inflammatory mechanisms
[9][142]. Investigating the mechanistic basis for these bioactive metabolites’ actions will aid in
ou
r understanding of cancer biology (
Table 1). Besides, here is also included an extensive number of scientific outcomes, where QC is a vital therapeutic agent in cancer stem cells and mitigates the devastating role of cancers, and the results are represented in
Table 2.
Figure 1.
The anticancer pathways and mechanisms of action induced by quercetin.
2. Reactive Oxygen Species-Mediated Regulation of the MAPK/ERK1/2 Pathways
2. ROS-Mediated Regulation of the MAPK/ERK1/2 Pathways
Quercetin plays a crucial role in several signal transduction pathways. Herein, it directly activates the MAPK/ERK-mediated pathways, leading to the apoptosis process and the mechanism discovered within the A549 cell lines of lung carcinoma
[10][69]. Besides, it induces the production of inflammatory and/or proinflammatory cytokines, and inhibits the formation of cyto-kinase LPS, which results in the suppression of iNOS by following the ERK, MAPK, as well as p38 mechanism
[11][143]. Basically, ERK1 or 2 is the major section of the MAPK cascade mechanism, these build up kinase family proteins namely MEK, Raf, and ERK1/2 that work successively
[12][144]. The active ERK 1 or 2 causes are reprogramming events linked to the expression of genes through the active phosphorylation process of various intracellular molecular target proteins and other transcription factors
[13][75].
Table 2.
Tabular representation of quercetin-mediated cancer stem cell killing with their mechanistic illustration.
3. Reactive Oxygen Species-Mediated Regulation of the p53 Pathway
3. ROS-Mediated Regulation of the p53 Pathway
The p53 pathway has been found to modulate cellular stress responses and to be vital in the direction of apoptosis in cancer. It is directly associated with MDM2 because of the complexities of p53, and is regulated at low levels by the regular proteasomal breakdown in unstressed cells
[46][176]. Similar to other cell signaling molecules, reactive oxygen species (ROS) function as an influencer in the activation of p53 through upstream signal transduction and also act as a downstream component to induce cell death (
Figure 2)
[47][106]. Quercetin, like other omnipresent bioactive plant flavonoids, has been demonstrated to reduce overall tumor growth and metastasis
[48][177]. Quercetin inhibited tumor cell proliferation by concurrently stimulating two divergent regulatory networks, p53 and NF-κB
[49][70]. An investigation reported that in A549 human lung cancer cells expressing wild-type p53, cell growth arrest and apoptosis were accelerated by quercetin considerably
[50][71].
Figure 2.
Mechanistic illustration of the function of quercetin in cancers cells by regulating ROS and p53-related pathways.
Another study found that while quercetin increased p53 phosphorylation, it did not increase p53 mRNA transcription. Quercetin increased p21 expression while suppressing cyclin D1 expression, favoring cell cycle arrest. In favor of apoptosis, the enhancement of the Bax/Bcl-2 ratio with this sort of therapy is also mediated by quercetin. Noticeably, the prevention of p53 mRNA degradation at the post-transcriptional stage occurred through quercetin-mediated signaling
[51][178].
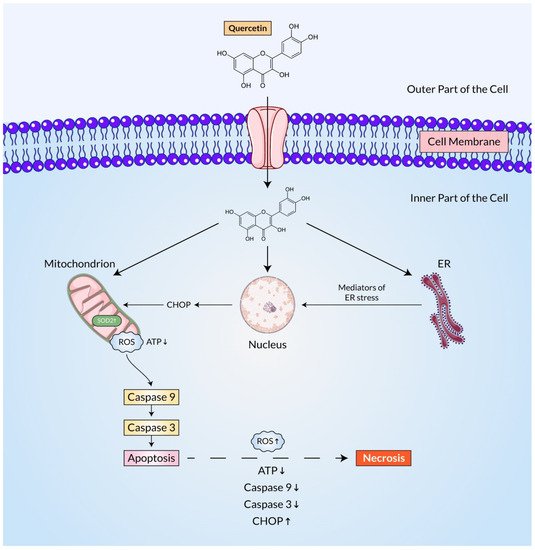
Lee, Yoon-Jin et al. showed that experiment on MM MSTO-211H cells, quercetin induced apoptotic cell death at low concentrations. According to the study, the up-regulation of p53 induced by quercetin can be visualized at both mRNA and protein levels without altering its ubiquitination as well as enhancing caspase-3/7 activity and procaspase-3 and PARP cleavages. The subset of cells in the G2/M phase that has been obstructed by the pan-caspase inhibitor escalated later in a quercetin concentration-dependent approach
[52][179]. Another study found that quercetin interacts with the human glioblastoma A172 as well as LBC3 cell lines, confirming increased ROS production, unregulated SOD1 and SOD2 expression, ATP depletion, and CHOP8 overexpression, and as a result, apoptosis of these brain cancer cell lines. They also reported enhanced caspase-9 expression and activity, supporting a mitochondrial apoptosis pathway
[53][180].
4. Reactive Oxygen Species-Mediated Regulation of the JAK/STAT and TRAIL Pathways
4. ROS-Mediated Regulation of the JAK/STAT and TRAIL Pathways
The JAK-STAT pathway is a well-characterized signaling pathway involved in the production of a variety of inflammatory and/or proinflammatory cytokines along with growth-controlling factors. The connection of ligand molecules to specific receptors leads to JAK mechanism activation that also improves the auto-phosphorylation process and activates the STAT signaling pathways
[54][181]. Several research studies revealed that the active form of mast cells directly increases the formation of Th2 cells such as cytokines, and at the same time reduces the secretion of cytokines mainly Th1. Similarly, mast cells regulate the expression of the JAK/STAT gene, which stimulates IL-13 development in the Th2 cell line
[55][182].
Quercetin can effectively inhibit the signaling pathways of JAK-STAT in various inflammatory conditions. Additionally, quercetin therapy of activated T-cells suppressed the phosphorylation of the TiK-2 (tyrosine kinase-2), JAK2, STAT-3,4 enzymes stimulated by interleukin-12, resulting in diminished T-cell proliferation, and Th1 variation
[56][183]. As a result, quercetin’s anti-inflammatory and anti-apoptotic properties play an important role in cancer prevention by modulating the TLR-2 (toll-like receptor-2) and JAK-2/STAT-3 pathways and significantly inhibiting STAT-3 tyrosine phosphorylation within the inflammatory cells
[57][72]. QC-mediated pretreatment of cholangiocarcinoma cells inhibited the JAK/STAT cascade pathway-mediated activation of iNOS and activation of the ICAM-1 (intercellular adhesion molecule-1). Additionally, QC inhibited the inflammatory cytokine IL-6 and interferon-alpha activation
[58][184].
5. Reactive Oxygen Species-Mediated Regulation of the AMPKα1/ASK1/p38 Pathways
5. ROS-Mediated Regulation of the AMPKα1/ASK1/p38 Pathways
Understanding the mechanisms of apoptotic pathways can lead to the development of effective cancer preventive and treatment options. Particularly, outside stimuli and mitogen-activated protein (MAP) kinases are two well-known intracellular signaling pathways that possess effective signals of the apoptosis transducing channel named the kinase cascade. Apoptosis signal-regulating kinase (ASK-1) is a redox sensor that belongs to the ROS-sensitive MAP kinase kinases family
[59][120]. The chemopreventive drug, quercetin, reported potent apoptosis induction in cancer cells mediated by ASK-1. Quercetin is responsible for free radical-induced cell death by the production of sufficient reactive oxygen species (ROS) though it has conventional anti-oxidation activity in the body. Through the apoptosis signal-regulating kinase (ASK)-1 and mitogen-activated protein kinase pathways,
rwe
searchers studied the regulation mechanism of quercetin-induced apoptosis
[60][118]. A therapeutic technique for cancer treatment is to induce apoptosis in cancer cells
[61][185]. Quercetin (3,30,40,5,7-pentahydroxyflavone) has a broad-spectrum of biological effects, along-with cancer prevention and tumor growth suppression
[62][186]. After scavenging the peroxyl radicals present, quercetin is transformed to quercetin-O-, a radical form that emerges to be one of the multiple pathways participating in the formation of ROS by quercetin
[60][118]. Due to the generation of quercetin radicals (quercetin-O•) throughout its interaction with peroxyl radicals, quercetin also enhances intracellular reactive oxygen species (ROS), inducing free radical-induced apoptosis via the ROS/AMPK1/ASK1/p38 and AMPK1/COX2 signaling pathways
[63][187]. AMPK1 and ASK1 are activated when the quantity of ROS produced increases in which AMPK1 and ASK1 activate p38 and boost the activity of several caspases
[62][186]. I
n t
he current study, it was reported that the quercetin-mediated apoptosis process is regulated by the activation of AMP-activated protein kinase (AMPK)/p38 MAPK signaling pathways and enhanced expression of sestrin 2. According to the findings, quercetin triggered apoptosis via raising the expression of sestrin 2 and creating intracellular reactive oxygen species (ROS). The activation of the AMPK/p38 signaling pathway by quercetin reported the induction of apoptosis, which was dependent on sestrin 2. Nevertheless, silencing sestrin 2 with short-interfering RNA (siRNA) demonstrated that quercetin had no effect on AMPK or p38 phosphorylation inside the cells where sestrin 2 was silenced
[61][185]. Another AMPK1 downstream target involved in Quercetin-induced apoptosis is COX-2
[64][8]. Quercetin radicals have also been reported to reduce the intracellular glutathione (GSH) pool in a concentration-mediated manner, as well as initiate apoptosis via mitochondrial depolarization. A research study demonstrated that quercetin exhibited interaction with the estrogen binding site type II
[63][187]. Apoptosis signal-regulating kinase 1 (ASK1) is a mitogen-activated protein (MAP) kinase that stimulates both the MKK3/MKK6–p38 MAP and MKK4/MKK7–JNK kinase pathways and is a key signaling route in many forms of stress-induced apoptosis. ASK1 has been discovered to have an important role in apoptosis triggered by stress, particularly oxidative stress, and endoplasmic reticulum (ER) stress
[65][188]. AMP-activated protein kinase (AMPK) appeared to be an important regulator of quercetin-mediated ASK1/p38 activation. When the activity of AMPKa1 was inhibited with a synthetic inhibitor or siRNA, quercetin-activated ASK1 was unable to promote p38 activity. As a result, it is thought that quercetin’s apoptotic effects are mediated by the ROS/AMPKa1/ASK1/p38 signaling pathway, and that AMPKa1 is required for ASK1-induced apoptosis. The alanine substitution of NR4A2’s oxidative stress-mediated phosphorylation sites reduces NR4A2’s cytoplasmic translocation and pro-necrosis activity, implying that p38-dependent phosphorylation of NR4A2 is a critical regulation to bestow the necrosis-inducing activity to NR4A2
[66][189]. These research outcomes imply that the ROS/AMPKa1/ASK1/p38 signaling pathway is involved in quercetin-induced apoptosis, and that AMPKa1 is a key regulator of ASK1
[60][118].
6. Reactive Oxygen Species-Mediated Regulation of the RAGE/PI3K/AKT/mTOR Axis
6. ROS-Mediated Regulation of the RAGE/PI3K/AKT/mTOR Axis
AKT signaling is required for the maintenance of normal physiological conditions. AKT signaling is frequently activated in cancer, which keeps the cellular microenvironment of tumors in a highly oxidative state, which is necessary for tumor development. As a result, antioxidants are hypothesized to possess anticancer effects through their ability to disrupt the tumor microenvironment. According to the findings of the study, quercetin (QC) prevented AKT signaling, which led to less cell survival, inflammation, and blood vessel growth in mice with lymphoma
[67][190]. In human pancreatic adenocarcinoma cells, QC enhanced cell death and chemosensitivity via the regulation of RAGE/PI3K/AKT/mTOR axis
[43][173].
Angiogenesis is a critical phase in cancer growth and spread because it allows the growing tumor to get oxygen and nutrients. In vitro, in vivo, and ex vivo antiangiogenic potential of quercetin reported that the progression of human prostate cancer was suppressed through the regulation of VEGFR-2-mediated Akt/mTOR/P70S6K signaling pathways
[68][191]. Quercetin treatment dramatically reduced the overexpression of pro-fibrotic factors (IL-6, IL-8, COL-1, COL-3, and LC3) as well as the increase in pro-fibrotic signaling mediators (mTOR and AKT) generated by LPS in WI-38 cells. Thus, nasogastric injection of QC significantly reversed an elevation in profibrotic markers (VEGF, IL-6, TGF, COL-1, and COL-3) and fibrotic signaling mediators (mTOR and Akt) in the rabbit tracheal stenosis research model, while also inactivating ATG5
[69][192]. QC blocked the PI3K/AKT/mTOR and STAT3 signaling pathways in primary effusion lymphoma cells, which induced cell death and autophagy
[70][193]. In addition, quercetin showed its anti-breast cancer effect via inhibiting the Akt/AMPK/mTOR signaling cascade
[71][194]. Because it inhibited the PI3K/Akt/mTOR signaling pathway and has been proven to reduce breast cancer stem cells (CD44+/CD24), it was identified as a candidate for the treatment of breast cancer
[72][195].
Q-6-C-b-D-glucopyranoside, a naturally occurring derivative of quercetin, demonstrated anti-prostate cancer action via blocking the Akt-mTOR pathway through the aryl hydrocarbon receptor
[73][196]. The generation of human breast cancer stem cells was inhibited due to the inhibition of the Notch1 and PI3K/Akt signaling pathways by quercetin-3-methyl ether
[74][197]. In addition, quercetin-3-methyl ether stopped the growth of cancer in the esophagus by blocking the Akt/mTOR/P70S6k and MAPK pathways, which are important for the growth of cancer
[75][198]. P53, Akt/mTOR pathway, and cancer cell growth were all downregulated in breast cancer cells when the vanadium quercetin complex is used, which is also associated with many apoptosis events
[76][199]. On the contrary, Burkitt’s lymphoma cells are significantly more susceptible to death because of quercetin’s induction of a drop in c-Myc expression in addition to PI3K/AKT/mTOR signaling
[77][200]. Quercetin inhibited glycolysis via activating the Akt-mTOR pathway, resulting in the activation of autophagy, hence inhibiting the migration of breast cancer cells
[78][201]. In glycolysis-mediated HCC cells, it suppressed the hexokinase 2 and the Akt-mTOR pathway, which are involved in glycolysis-dependent cell proliferation
[79][202]. Quercetin has been shown to suppress the Akt-mTOR pathway and hypoxia-induced factor 1 signaling pathway in gastric cancer cells, resulting in preventative autophagy
[80][203].
Furthermore, quercetin possessed strong synergistic activity in combination with anti-sense oligo gene therapy (suppressing snail gene expression), it significantly inhibited the progression of renal cell carcinoma Caki-2 by the regulation of Akt/mTOR/ERK1/2 signaling pathways
[81][204]. Quercetin nanoparticles inhibited the AKT/ERK/caspase-3 signaling pathway to induce autophagy and apoptosis in human neuroglioma cell lines in vitro and in vivo research models
[82][205]. In breast cancer cell lines (MCF-7 and MDA-MB-231), quercetin coupled with gold nanoparticles promoted apoptosis by inhibiting the EGFR/P13K/Akt-mediated pathway
[42][172].
7. Reactive Oxygen Species-Mediated Regulation of the HMGB1 and NF-κB Pathways
7. ROS-Mediated Regulation of the HMGB1 and NF-κB Pathways
The HMGB1 (high mobility group box 1) protein that is exposed to the outside environment has the potential to trigger the production of TNF (tumor necrosis factor), IL-1, and other inflammatory cytokines from monocytes
[83][206]. QC enhances the suppression of HMGB1-induced TNF and IL-1 mRNA production, indicating that it transmits signals inside cells that modulate the action of proinflammatory cytokines
[84][207]. In HMGB1-induced gene expression, the activation of MAPK is important for the release of inflammatory cytokines such as IL-1 and TNF from neutrophils, macrophages, and endothelial cells. The release of cytokines induced by HMGB1 partially interferes with MAPK pathways in which the p38 phosphorylation or C-Junction of N-terminal kinases with extracellular signals regulate kinases in macrophages in a time-dependent manner by HMGB1 or LPS. Here, such a bioactive compound significantly inhibits the activity of each kinase enzyme by maintaining the phosphorylation process
[85][208]. Additionally, a recent study reported that QC can easily modulate the signaling proteins (i.e., Cox-2, NF-κB) that may lead to the apoptosis process, and furthermore, suppress anti-apoptotic proteins, e.g., Bcl-2 as well as Bcl-xL, up-regulating Bax with several pro-apoptotic proteins
[86][87][209,210]. In addition to activating MAPK, the nuclear factor-κB (NF-κB) pathway also includes HMGB1-induced cellular expression, and NF-κB-dependent gene expression is of significant importance when exposing cytokine
[88][89][211,212]. Through their interaction with the IκBα (members of the IκB family), subunits of NF-κB (p50 as well as p65) take place in cells as inactive trimers within the cell cytosol
[90][213]. Interestingly, QC suppresses the degradation of IκBα factor and NF-κB p65 in a significant manner. As a result, following stimulation with the following p65, HMGB1 or LPS, the critical mediator of NF-κB factor will become available to be regulated nuclear NF-κB genes and the compound QC most effectively inhibits the nuclear localization process
[13][75].
8. Reactive Oxygen Species-Mediated Regulation of the Nrf2-Induced Phase II Enzyme and Signaling Pathways
8. ROS-Mediated Regulation of the Nrf2-Induced Phase II Enzyme and Signaling Pathways
Carcinogenesis can be suppressed through inductions from Phase II enzymes, including the GST (glutathione S transferase), NAD(P)H/NO (quinone oxidoreductase), UDP-GLT (UDP-glucuronosyl transferases), and HO-1 (heme oxygenase-1)
[91][92][214,215]. The genes of such enzymes bearing the ARE (antioxidant replication elements), which are strictly regulated by Nrf2 nuclear erythroid, and which, in turn, are associated by Keap-1 (Kelch-like CEH-associated protein-1), therefore, an Nrf2 represent, also encourage their degradation through the path of the ubicin-dependent proteasome
[93][94][95][216,217,218]. When cells are treated with ARE-mediated stimulants such as QC, the complex formation of “Nrf2-Keap1” dissociates, allowing the Nrf2 factor to be translocated from the cytosol to the nuclear part of the cell. By binding to ARE and subsequently forms a heterodimer structure with other transcription factors (TFs), which simultaneously promotes the transcription of phase II enzyme genes.
[96][97][219,220]. QC has also been demonstrated to stimulate ARE binding activity and regulate the mRNA expression level of NQO1, the total mechanism that occurred in the HepG2 cell line in a dose-dependent manner
[97][220]. In addition, QC regulated the prohibition of Nrf-2 protein degradation and maintained the reduced amount of posttranslational products of Keap-1 proteins without any impairment of the complex structure dissociation like- “Keap-1-Nrf2”
[97][220]. A cross-mediated increase in enzymes of phase II Caco-2 adenocarcinoma with duodenum adenocarcinoma of HuTu 80 cells in human CRAC (colorectal adenocarcinoma) was shown
[98][221]. The effect of QC on nuclear translocation of Nrf-2 in a time-dependent manner, and increased expression level in HepG2, MgM (malignant mesothelioma) MSTO-211H, and H2452 cells at mRNA and protein quantity has been reported recently
[99][222]. Moreover, the therapeutic efficacy of QC has also been defined in rat models through the activation of Nrf-2/HO-1 against high glucose-induced damage
[100][223]. Until now, little is known about the precise biochemical pathways by which QC triggers Nof2-based gene expression. Nevertheless, evidence suggests that the improvement of diverse signal transduction cascades, along with the MAPK, modulates cross-induced interpretation of transcriptional genes (MAPK). The p38 and ERK-generated translocation of Nrf2 to nuclei between MAPK signal pathways were shown to be responsible for the successive initiation and activity of HO-1 expression
[101][102][103][224,225,226]. Indeed, QC (50 µM) protected RAW264.7 macrophages from the induction of apoptosis by up-regulating phase II enzymes, which include HO-1, via ERK pathway-dependent mechanisms
[102][225]. Similarly, cross-treated human hepatocytes were mentioned in MAPK HO-1-dependent upregulation and successive preservation against ethanol-induced oxidative damage
[103][226].