Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ron Firestein and Version 2 by Catherine Yang.
The Wnt/β-catenin signaling pathway exerts integral roles in embryogenesis and adult homeostasis. Aberrant activation of the pathway is implicated in growth-associated diseases and cancers, especially as a key driver in the initiation and progression of colorectal cancer (CRC).
- Wnt/β-catenin signaling pathway
- transcriptional regulation
- epigenetic regulation
- colorectal cancer
1. Introduction
Wnt signaling is a growth control pathway that can regulate many biological processes ranging from development and evolution to adult homeostasis. Wnt signaling includes two branches, canonical (β-catenin-dependent activity) and non-canonical (β-catenin-independent activity) Wnt pathways. β-catenin is composed of a central region, including twelve imperfect Armadillo repeats, which are flanked by different domains in N- and C-terminus, respectively [1]. It acts as a crucial nuclear effector of the canonical Wnt pathway. WNT proteins are important intracellular ligands that stimulate the canonical Wnt pathway. In the absence of a WNT ligand, cytoplasmic β-catenin is regulated by the destruction complex, mainly containing Adenomatous polyposis coli (APC), Axis inhibition protein (AXIN), Glycogen synthase kinase 3 (GSK3) and Casein kinase 1 (CK1) for final degradation [2][3][4][2,3,4]. The destruction complex induces continuous elimination of β-catenin, thus impeding its nuclear transport, so that expression of Wnt target genes is switched off. WNT ligand binding to the FZD-LRP5/6 receptor complexes stabilises cytoplasmic β-catenin via either inhibition of phosphorylation of β-catenin combined with the disassembly of the destruction complex [5][6][7][8][5,6,7,8] or inactivation of ubiquitination and prosomal degradation of β-catenin in the intact destruction complex [9]. Once stabilised, cytoplasmic β-catenin accumulates and translocates to the nucleus where it binds to transcription factors, such as the TCF/LEF family [10], together with the recruitment of different nuclear regulators, to activate downstream transcriptional cascades of the Wnt pathway (Figure 1). Several mutations in the pathway can cause ligand-independent β-catenin stabilisation, which, thus, contributes to oncogenic β-catenin-regulated transcriptional activity [11].
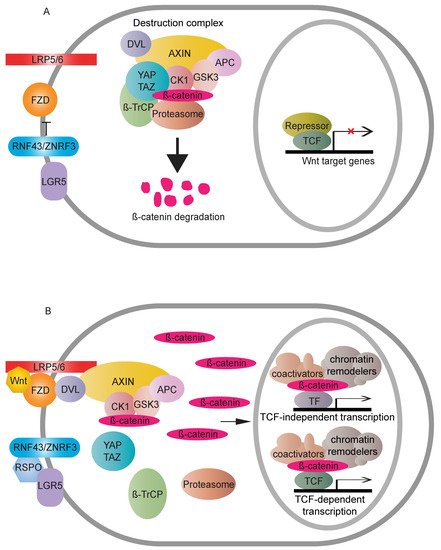
Figure 1. Overview of the Wnt canonical (β-catenin-dependent) pathway. (A) In the absence of a Wnt signal, cytoplasmic β-catenin is degraded by the destruction complex. RNF43/ZNRF3 targets FZD to antagonise Wnt signalling. The accumulative result is a transcriptional “off” state. (B) Wnt binding causes dimerisation of FZD and LRP5/6. The receptor complex recruits the destruction complex to the cell membrane, and DVL assists the interactions between LRP5/6 and AXIN. Cytoplasmic β-catenin, thus, can be relieved from proteasome-dependent degradation. Stable β-catenin accumulates in the cytoplasm, followed by nuclear transport. There, β-catenin displaces repressive complexes and forms active complexes through associations with transcription factors and recruitment of coactivators and chromatin remodelers to upregulate Wnt responsive targets. RNF43/ZNRF3′s inhibition on the Wnt pathway can be counteracted by binding of RSPO to LGR5.
Moreover, many regulators have been identified which control β-catenin’s subcellular localisation, nuclear abundance or transcriptional activity. Negative feedback loops, comprising AXIN or RNF43/ZNRF3, which are themselves Wnt downstream targets, exemplify the necessity of tight homeostatic control. The pathway is further complicated by the presence of another regulatory axis, LGR5-RSPO-RNF43/ZNRF3 [12][13][14][12,13,14]. Together, the involvement of multiple regulatory factors that can translate Wnt signals into different downstream readouts provides an explanation for the diversity and complexity of Wnt signalling (Figure 2).
Figure 2.
Multidimensional regulators in the Wnt signalling pathway. The hallmark determinants of β-catenin transcriptional output are depicted.
2. Mediator Kinase of β-Catenin-Mediated Transcriptional Output
The Mediator complex acts as a crucial component in transcription regulation, in light of its ability to transduce signals from pathway-specific transcription factors (TFs) to regulate RNA polymerase II (RNAPII) activity [15][16][117,118]. Depending on its functional configuration, the Mediator complex consists of 25 to 30 proteins, which can be classified into four submodules—head, middle, tail and Mediator kinase (Cyclin-dependent kinase 8 (CDK8), 19) module. The CDK8 module contains CDK8 (or its CDK19 paralog), Cyclin C (encoded by CCNC), MED12 and MED13 subunits. CDK19, a highly conserved paralog of CDK8 in vertebrates, shares conservation in the kinase and cyclin binding domain but is divergent at the C-terminal tail [17][119]. CDK8 and CDK19 are incorporated into the Mediator kinase module by way of binding MED12 in a mutually exclusive manner. Cyclin-dependent kinase (CDK) proteins mainly consist of two types of members, cell cycle progression CDKs, such as CDK1, CDK2 and CDK4/6, and transcriptional CDKs, such as CDK7, CDK9 and CDK8/19 [18][120]. These transcriptional CDKs complex with their cyclin subunit to phosphorylate the C-terminal domain (CTD) of RNAPII, regulating transcription initiation, elongation and mRNA processing [19][121]. Unlike CDK7 and CDK9, CDK8/19 activity is dispensable for general transcription regulation and normal cell growth. Recently, the CDK8 kinase function has been linked to an “activation helix” placed by MED12 close to the CDK8 T-loop [20][122]. CDK8/19 acts downstream of several transcription factors, such as HIF-1α [21][123] and NFκB [22][124], to transcribe their target genes. In addition, apart from its action on RNAPII, CDK8 phosphorylates some TFs, such as SMAD, to regulate β/BMP signalling [23][125]. CDK8/19 is associated with diverse TFs to selectively enhance the expression of silent genes, implying their regulation is in a context-specific manner.
CDK8/19 has been implicated in several cancers, especially those induced by dysregulated gene expression rather than mutations [24][126]. It has been more than a decade since CDK8 was identified as an oncoprotein in colon cancer. CDK8 was found to act, in a kinase-dependent manner, as a driver of β-catenin-dependent transcription and colorectal cancer proliferation [25][127]. The underlying molecular mechanisms of how CDK8 regulates β-catenin-dependent transcription are less-well defined, especially considering that the critical kinase substrates of CDK8 that mediate this activity are yet to be identified. Converging genetic studies have demonstrated that the ability of CDK8 to drive β-catenin-regulated gene expression is likely Mediator complex dependent. Aside from CDK8, Mediator kinase submodule proteins MED12 and MED13 have been implicated in recruiting Mediator to Wnt-directed target genes and regulating β-catenin transcriptional output [26][27][128,129]. Furthermore, repressing the CDK8 co-factor, Cyclin C, produces similar influences on β-catenin-mediated transcription as the inhibition of CDK8 [25][127]. Nevertheless, it has also been proposed that CDK8 can exert its effects on β-catenin indirectly via potentially Mediator-independent mechanisms. Morris et al. suggested pRB and CDK8 can counteract the inhibition of E2F1 on β-catenin-regulated transcription to enhance the expression of Wnt/β-catenin genes [28][130]. Indeed, chromosomal copy number gains in both CDK8 and RB1 loci on 13q in colorectal cancer cells provide a clear explanation of how they select optimal conditions to antagonise E2F1 suppressions [25][28][127,130]. E2F1 can post-translationally degrade β-catenin [28][130] and can activate ICAT which disrupts β-catenin-TCF/LEF complex [29][131]. The physical interaction between CDK8 and E2F1 at the promoters of β-catenin targets, such as MYC, provides a basis for understanding how Mediator kinase may enhance β-catenin transcriptional output [28][130]. Subsequent studies have further demonstrated that CDK8 phosphorylates Ser375 on E2F1 in colon cancer cells. This phosphorylation alleviates the E2F1 repressive effects on β-catenin-associated transcription [30][132]. Together, these data imply that E2F1 could be one of the crucial substrates of CDK8 to enable its regulation on Wnt/β-catenin transcriptional output. Moreover, a recent study identifies YAP as a novel substrate of CDK8. In Hippo signalling, YAP is phosphorylated directly by CDK8 to induce its function, and the phosphorylated YAP can be regulated further by Zyxin, a zinc-binding phosphoprotein, to promote cell proliferation and migration in CRC [31][133]. In conclusion, CDK8 kinase activity plays an essential role in driving oncogenic events. It can activate different substrates that are associated with intestinal neoplasia and tumour progression. Accordingly, it is quite possible that therapeutic interventions targeting its kinase activity could have clinical value in the treatment of CRC.
3. Chromatin States Regulate β-Catenin-Mediated Transcriptional Output
Chromatin remodeling is an essential step for transcription regulation. The nucleosome, the basic subunit of chromatin, is composed of histones H2A, H2B, H3, H4 and 146 base pairs (bp) of DNA. These histone tails undergo diverse post-translational modifications, such as acetylation, methylation, phosphorylation, ubiquitination, sumoylation and ADP ribosylation. Such modifications can increase or decrease the accessibility of DNA for transcriptional regulators to coordinate gene expression [32][33][34][134,135,136]. Histone acetyltransferases (HATs) and deacetylases (HDACs) are associated with enhanced and decreased acetylation, which mark gene activation and repression, respectively [35][137]. Furthermore, histone methylation can both alter chromatin accessibility and epigenetically activate or inactivate gene expression. The dynamic balance between histone methylation and demethylation is mediated by histone methyltransferases (HMTs) and demethylases (HDMs), respectively [36][138]. In addition to histone modification, chromatin remodeling is also an important epigenetic event. ATP-dependent remodeling complexes can alter the location and configuration of the nucleosome, and the changed chromatin can participate in either gene activation or inhibition [35][137]. Epigenetic regulation is involved in various events, including transcription, replication, repair and genome stability. Lastly, chromatin modifiers have been reported to have essential influences on cancer etiology [37][38][39][139,140,141]. In Wnt signalling, various chromatin modifiers have been identified as activators or repressors of Wnt/β-catenin transcriptional outputs (Figure 35).
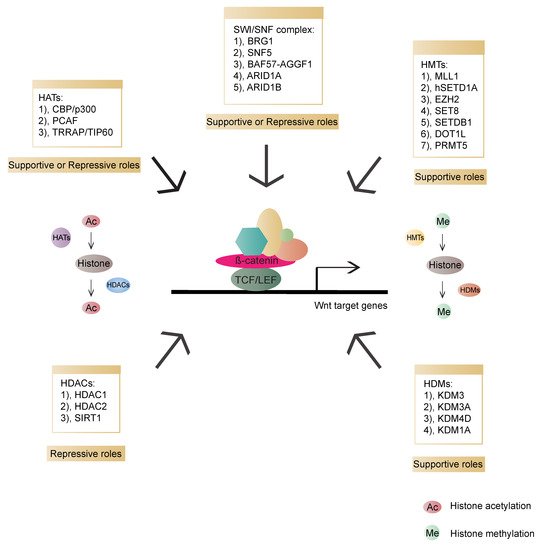
Figure 35. Overview of chromatin modifiers involved in β-catenin-dependent transcriptional regulation and their roles in the Wnt pathway. HATs (histone acetyltransferases, transferring acetyl groups to histones): CBP (CREB-binding protein)/p300, PCAF (p300/CBP-associated factor), TRRAP (Transformation/transcription domain-associated protein)/TIP60; HDACs (Histone deacetylase, removing acetyl groups from histones): HDAC1 (Histone deacetylase 1), HDAC2 (Histone deacetylase 2), SIRT1 (NAD-dependent deacetylase sirtuin-1); HMTs (Histone methyltransferases, transferring methyl groups to histones): MLL1 (Mixed-lineage leukemia 1 or Histone-lysine N-methyltransferase 2A), hSETD1A (human SET domain containing protein 1A), EZH2 (Enhancer of zeste 2 polycomb repressive complex 2 subunit), SET8 (SET domain-containing protein 8), SETDB1 (SET domain bifurcated 1), DOT1L (DOT1 like histone lysine methyltransferase), PRMT5 (Protein arginine N-methyltransferase 5); HDMs (Histone demethylases, removing methyl groups from histones): KDM3 (Lysine-specific demethylase 3), KDM3A (Lysine demethylase 3A), KDM4D (Lysine-specific demethylase 4D), KDM1A (Lysine demethylase 1A); SWI/SNF: BRG1 (Transcription activator BRG1), SNF5 (BRG1-associated factor 47), BAF57 (BRG1-associated factor 57), AGGF1 (Angiogenic factor with G patch and FHA domains 1), ARID1A (AT-rich interactive domain-containing protein 1A), ARID1B (AT-rich interactive domain-containing protein 1B).
6. Therapeutic Strategy
The clinical implications of targeting the Wnt signalling pathway has been recognised for decades. Nevertheless, this has not been translated into the clinic for reasons detailed below. Dysregulated Wnt pathway can be targeted at three different levels: the Wnt-receptor complex, the cytoplasmic destruction complex and the nuclear β-catenin-TCF/LEF complex. Regulators involved in downstream transcriptional levels emerge as the most attractive targets for drug development. The implications of APC loss in the vast majority of CRC make inhibitors targeting Wnt signalling components upstream of the destruction complex ineffective. One exception is CRCs harbouring RSPO2/3 fusions [40][209]. While accounting for only 1–3% of CRC cases, the ability to therapeutically target RSPO2/3 fusion driven CRCs offers a proof of concept evidence that specific cancer targeting for the Wnt pathway can be achieved [41][210]. Recent studies in human colon organoids and in in silico genetic interaction analysis provide a model of how homeostatic and oncogenic Wnt responses can be untangled and stratified [42][43][211,212]. Diverse transcriptional regulators provide potential avenues to identify those only contributing to tumourigenesis. This diversity within this growing toolbox of targets offers opportunities to design rational therapies that will specifically target oncogenic signalling with minimal normal toxicities.
Disrupting protein–protein interactions in transcriptional complexes opens a new therapeutic window for anti-cancer drug development, although the transient nature of some interactions hinders effective drug development. Currently, most of the inhibitors targeting transcriptional level in Wnt pathway attempt to disrupt β-catenin-TCF4 interactions [44][45][46][47][48][49][213,214,215,216,217,218]. In addition, inhibitors interfering with the interaction between β-catenin and CBP may be suggested as attractive candidates [50][51][52][53][219,220,221,222]. Recently, in APC-deficient models, it was demonstrated that abrogation of BCL9/9L cannot affect intestinal homeostasis, but can negatively influence intestinal neoplasia, thus highlighting the therapeutic potential of targeting the interaction between β-catenin and BCL9/9L [54][55][223,224]. Kinase inhibitors are a mainstay of cancer therapies. The ability of a specific CDK8/19 kinase inhibitor, CCT251545, inhibits Wnt pathway-induced tumours in mouse models [56][225]. Similarly, another kinase, YES1, implicated in YAP dependent β-catenin transcriptional output, can be inhibited by CH6953755 to restrict YES1-YAP1-dependent tumour growth [57][58][94,226]. Lastly, NSC48300, a pharmacologic inhibitor for Taspase1, can effectively inactivate breast and brain tumour growth via preventing the formation of a mature MLL1 complex [59][227]. Current inhibitors and their drug development status are summarised in Table 12.
Table 12.
Summary of potential therapeutic strategies and inhibitors targeting dysregulated Wnt pathway.
| Therapeutic Strategies | Targets | Inhibitors | Clinical Stage |
|---|---|---|---|
| Targeting the Wnt-receptor complex | Porcupine (PORCN) (important for Wnt secretion) |
LGK974 [60][228]; ETC-159 [61][229]; | LGK974; ETC-159 |
| WNT-FZD | Vantictumab (OMP-18R5) [62][230]; Ipafricept (OMP-54F28) [63][231]; SFRP-1 [64][232]; Foxy-5 [65][233]; OTSA-101 [66][234] | OMP-18R5; OMP-54F28; Foxy-5; OTSA-101 | |
| LRP | Niclosamide [67][235]; Salinomycin [68][236]; Monensin [69][237] | Niclosamide (FDA approved) | |
| RSPO | OMP-131R10 [70][238] | OMP-131R10 | |
| Targeting the cytoplasmic destruction complex | FZD-DVL | 3289-8625 [71][239]; FJ9 [72][240]; NSC668036 [73][241] | None |
| Tankyrase (an inhibitor of AXIN) |
XAV939 [74][242]; IWR-1 [75][243]; JW55/67/64 [76][77][244,245]; JW74 [78][246]; G007-LK [79][247]; MSC2504877 [80][248]; RK-287107 [81][249]; NVP-TNKS656 [82][250] | None | |
| CK1 | Pyrvinium [83][251] | Pyrvinium (FDA approved) | |
| GSK3 | LY2090314 [84][252] | LY2090314 | |
| Targeting the nuclear β-catenin-TCF/LEF complex | Β-catenin | MSAB [85][253] | None |
| Β-catenin-TCF4 | PKF115-584 and CGP049090; iCRT3, 5, 14; NC043; BC21; LF3; HI-B1 [44][45][46][47][48][49][213,214,215,216,217,218] | None | |
| Β-catenin-CBP | ICG-001 [50][51][52][219,220,221]; PRI-724 [86][254] | PRI-724 | |
| Β-catenin-BCL9/9L | SAH-BCL9 [87][255]; hsBCL9CT [88][256] | None | |
| Β-catenin-TRIB3 | P2-T3A6 [89][72] | None | |
| TNIK | N5355 [90][257] | None | |
| CDK8/CDK19 | CCT251545 [56][225] | None |
Table 12 summarises the main therapeutic strategies, targets and inhibitors of the Wnt pathway in cancer. The clinical status of each inhibitor is noted. Of note, niclosamide and pyrvinium have been approved by FDA.
Epigenetic modifications are reversible, thus attracting extensive attention to targeting abnormal epigenetic events so as to revert the cancer phenotype to a normal state. Despite promising therapeutic potential in hematopoietic malignancies, it is not clear why epigenetic inhibitors exert less efficacy in solid tumours. Currently, drugs targeting epigenetic regulators directly in CRC are missing. Resistant mechanisms need to be explored to provide a better opportunity for the development of epigenetic drugs in solid cancers.