Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alin Grig Mihis and Version 2 by Jason Zhu.
ATP-binding cassette subfamily G and tubulin pharmacological mechanisms decrease the effectiveness of anticancer drugs by modulating drug absorption and by creating tubulin assembly through polymerization. A series of natural and synthetic chalcones have been reported to have very good anticancer activity, with a half-maximal inhibitory concentration lower than 1 µM. By modulation, it is observed in case of the first mechanism that methoxy substituents on the aromatic cycle of acetophenone residue and substitution of phenyl nucleus by a heterocycle and by methoxy or hydroxyl groups have a positive impact. To inhibit tubulin, compounds bind to colchicine binding site. Presence of methoxy groups, amino groups or heterocyclic substituents increase activity.
- chalcones
- molecular hybridization
- ABCG2 cassette
1. Introduction
Cancer is characterized by uncontrolled, progressive, rapid and pathological cell proliferation and is one of the most aggressive diseases in the world [1][2][1,2]. This condition is a correlation of pathologies characterized by uncontrolled and abnormal cell growth, which are based on gene mutations [3][4][3,4]. The altered activity of some transcription factors determines the appearance of many types of cancers [5]. Heterogeneity of cancer cells targets is associated eith genomic instability, gene variation in tumor formation and ability of tumor cells to have excessive phenotypic variations [6]. This has important clinical consequences, as tumor formations with complex subclonal structures can be aggressive and have the ability to initiate resistance to therapy and form metastases [7][8][7,8]. The formation of metastases to distant organs is often incurable and is a determining factor in the death of cancer patients, which in these cases, is frequently associated with dysfunctions of vital organs [9][10][9,10]. Cancer cells have aberrant redox homeostasis, reactive oxygen (ROS) species by being pro-tumorigenic and, at high levels of ROS, have cytotoxic activity [11][12][11,12]. Due to aberrant redox homeostasis in tumor cells and their microenvironment, these reactive species are insoluble in carcinogenesis, but also in anticancer therapy. Numerous studies have shown that the role of ROS in malignant cells, positive or negative, depends on factors such as tumor type, cancer stage, therapeutic strategies, duration of cell exposure to ROS, specificity and ROS level [13][14][13,14]. The increased number and clinical importance of molecular biomarkers in usual practice allow anticancer therapies to have an increased specificity on a particular genetic complex in tumor formation. The disadvantages of these biomarkers are related to the expected duration of analyses and the need to take samples from tissues [15]. There are four major types of approaches available for cancer treatment (radiotherapy, surgery, immunotherapy and drug therapy) [16][17][18][19][20][16,17,18,19,20]. Less than a quarter of patients with common cancers receive appropriate therapy [21]. Early diagnosis of cancer provides the opportunity to choose the appropriate therapeutic intervention. Among methods used for early diagnosis are tumor markers and imaging techniques, such as computed tomography, magnetic resonance imaging, position emission tomography and ultrasound scanning endoscopy (cytogenetic and cell genetics screening) and so forth [22]. Standard treatment for most cancers includes chemotherapeutic agents. The response to this type of therapy varies for each patient, especially in case of poorly characterized cancers. Tumor heterogeneity, the presence of cancer stem cells and the plasticity of these cells indicate the need to use combination therapies and/or targeted therapies [23]. Platinum compounds are a major component of chemotherapy for various types of cancer. Three platinum compounds used in anticancer therapy-cisplatin, carboplatin and oxaliplatin-have the ability to distort the normal functioning of DNA (deoxyribonucleic acid) by generating monoadducts [24]. Unlike chemotherapy, targeted therapy is considered more effective and safer for the treatment of cancer [25]. In recent years, immunotherapy has become a major alternative to conventional anticancer therapy, which aims to increase the quality of life of patients and prolong life [26]. Immunotherapy, ideal for fighting cancer, acts on the immune system to eradicate malignant cells [27]. There are two therapeutic directions of immunotherapy: blocking immune checkpoints by antibodies that block immune system inhibitory receptors in tumors, and adoptive cell therapy by which T cells are designed to express chimeric antigenic receptors [28][29][30][31][28,29,30,31]. It is also known that most recently approved targeted anticancer therapies are specific for coding oncoproteins for mutant somatic genes. These agents include proteins essential for the maintenance of specific cell descendants or proteins that inhibit immune responses, such as protein 1 of programmed cell death (PD-1) [32]. Immunotherapy, based on immunostimulatory monoclonal antibodies, explains the involvement of an immune response in anticancer therapy. PD1/PDL1 blockade demonstrates the importance of targeted immune mechanisms in tumor formation, by which T cell response in tumor formation is rehabilitated [33]. The most common problem with antitumor therapy is resistance to therapy [34][35][36][37][38][39][40][41][34,35,36,37,38,39,40,41]. Some cancer cells initiate resistance to many chemotherapeutic agents, others acquire this characteristic through mutations in the process of carcinogenesis [42]. This is correlated with cassette-binding ATP (ABC) transporters, which are P-glycoprotein (P-gP/ABCB1), multidrug resistance protein 1 (MRP1/ABCC1) and breast cancer resistance protein (BCRP/ABCG2). One method of treatment is the elimination of these resistant cancer cells, using multidrug ABC transporters [43][44][45][46][47][48][49][50][51][43,44,45,46,47,48,49,50,51].
2. Flavonoids
Flavonoids are the largest class of low-molecular-weight polyphenolic secondary metabolites with a benzopyron residue (C6-C3-C6) in the molecule. They consist of two aromatic rings joined by a bridge of three carbon atoms that form an oxygenated heterocycle [52][53][54][55][56][57][54,55,56,57,58,59]. Over 10,000 flavonoids have been described, many of which are present in large quantities in fruits and vegetables [58][60]. Flavonoids are biosynthesized by phenylpropanoid, starting from phenylalanine [59][61]. Chalcone synthetase is the first enzyme in flavonoid biosynthesis and catalyzes the formation of chalcones from one molecule of p-coumaroyl-CoA and three molecules of malonyl-CoA [60][62]. Depending on the degree of oxidation of the central nucleus, flavonoids are classified into flavones, flavanones, flavonols (proanthocyanidins), dihydroflavonols, isoflavones, isoflavanones, chalcones, aurones, anthocyanidins and tannins [61][62][63][64][63,64,65,66]. Flavonoid compounds are associated with a number of biological activities, their antioxidant, anticancer, anti-inflammatory, antidiabetic, antimicrobial, antidyslipidemic, neuroprotective, antiosteoporotic, cytoprotective, hepatoprotective, vasoprotective properties being much studied [65][66][67][68][69][70][71][72][73][67,68,69,70,71,72,73,74,75]. Flavonoids are known to decrease cardiovascular risk and cancer-associated mortality [74][76]. Experimental studies also show that dietary flavonoids interact with rhodopsin and modulate the functions of visual pigments [75][77]. The structural diversity of flavonoids is associated with chemical changes such as hydroxylation, methylation, acylation, glycosylation, etc. In plants, glycosylation is the most common modification of flavonoids, being responsible for increasing solubility and stability, maintaining bioactivity, regulating transport and accumulating and/or decreasing toxicity of these compounds [76][78]. By methylation of hydroxy groups of flavonoids, compounds with superior stability, a longer metabolism time, a favorable transport rate and, implicitly, a better absorption in target tissues are obtained [77][79]. For example, hesperidin (a natural flavanone, Compound 1) has low water solubility and has low absorption in the small intestine. By methylation of hesperidin in an alkaline medium, hesperidin methyl chalcone (Compound 2) is formed, a compound with superior water solubility and favorable intestinal absorption, bioavailability and tissue distribution [78][80]. In general, metallic complex combinations of flavonoids have superior cytotoxic, anti-inflammatory and redox properties [79][81]. Flavonoid compounds are associated with a number of biological activities, their antioxidant, anticancer, anti-inflammatory, antidiabetic, antimicrobial, antidyslipidemic, neuroprotective, antiosteoporotic, cytoprotective, hepatoprotective, vasoprotective properties being much studied [65][66][67][68][69][70][71][72][73][67,68,69,70,71,72,73,74,75]. Flavonoids are known to decrease cardiovascular risk and cancer-associated mortality [74][76]. Experimental studies also show that dietary flavonoids interact with rhodopsin and modulate the functions of visual pigments [75][77]. The structural diversity of flavonoids is associated with chemical changes such as hydroxylation, methylation, acylation, glycosylation, etc. In plants, glycosylation is the most common modification of flavonoids, being responsible for increasing solubility and stability, maintaining bioactivity, regulating transport and accumulating and/or decreasing toxicity of these compounds [76][78]. By methylation of hydroxy groups of flavonoids, compounds with superior stability, a longer metabolism time, a favorable transport rate and, implicitly, a better absorption in target tissues are obtained [77][79]. For example, hesperidin (a natural flavanone, Compound 1) has low water solubility and has low absorption in the small intestine. By methylation of hesperidin in an alkaline medium, hesperidin methyl chalcone (Compound 2) is formed, a compound with superior water solubility and favorable intestinal absorption, bioavailability and tissue distribution [78][80]. In general, metallic complex combinations of flavonoids have superior cytotoxic, anti-inflammatory and redox properties [79][81].2.1. Chalcones
Compounds with an enone system in molecules continue to be of particular interest due to their simple chemistry, the ease with which they are obtained by chemical synthesis, their biological importance and the possibility of being precursors in organic chemistry [80][82]. Chalcones (chalconoids, 1,3-diaril-2-propen-1-ones) are natural and synthetic compounds in which aromatic residues are joined by an α, β-unsaturated electrophiles chain of three carbon atoms [64][81][82][83][84][85][86][66,83,84,85,86,87,88]. Reactive hydrogens from the basic structure of chalcones determine the possibility of these compounds being structurally modified in order to obtain a large number of derivatives [87][89]. Chalcones are precursors for many heterocyclic compounds such as cyanopyridines, 2-pyrazolines, pyrazoles, isoxazoles, pyrimidine-2-ones, flavanones and flavones and so forth [88][89][90,91]. Data from literature indicate that molecules with a 1,3-diaryl-2-propen-1-one residue have numerous biological activities and, for this reason, are of particular pharmacological importance [90][91][92,93]. Among the most important biological activities of chalcones are antitumor, antibacterial, anti-inflammatory, antifungal, cytotoxic, antiviral, antimalarial, antituberculous, antiparasitic, antiulcer, analgesic, antipyretic, antihepatotoxic, etc., properties [92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120]. Chalcones have an extended conjugate system, which facilitates their binding to bioactive molecules, such as enzymes and DNA [119][121]. For example, chalcones are active on some enzymes such as acetylcholinesterase and carbonic anhydrase, being active in glaucoma, obesity, osteoporosis, epilepsy, Parkinson’s and Alzheimer’s [120][122]. The presence of α, β-unsaturated carbonyl group, which acts as a Michael acceptor, facilitates the interaction of chalcones with sulfhydryl groups in cysteine residues or with thiol groups. This interaction is considered to be responsible for the numerous biological properties of these compounds [121][122][123,124]. The biological activity of chalcones is also strongly influenced by their chemical structure, especially by substituents on two aromatic residues. It has advantages related to its ability to regulate molecular pathways related to cancer, favorably influence apoptosis, metastases and the response to cellular stress. Numerous methoxychalcones with antitumor properties have been identified, the presence of the methoxy group being favorable for this activity [123][124][125][126][125,126,127,128]. Since chalcones are active in vitro and in vivo in cancers susceptible to therapy, as well as in case of resistant ones, these compounds represent an important skeleton for the identification of new anticancer agents. In addition, 1,3-diaryl-2-propen-1-ones represent an important class of natural small molecules with anticancer chemotherapeutic action [127][129]. For example, chalcones are important pharmacophores for many natural products, such as flavokawaina, millepachine and xanthohumol (XN) (Compounds 3–7) [128][130]. XN (Compound 7), a prenylated chalcone from female hop inflorescences, inhibits the growth of MCF-7 (human breast adenocarcinoma cancer cells) and induces their apoptosis. Prenylchalcone is also effective in hepatocellular carcinoma and prostate cancer [129][131]. Mechanisms of the anticancer activity of XN include inhibition of initiation and progression of carcinogenesis, induction of apoptosis, and inhibition of angiogenesis [130][131][132,133]. Natural compounds with 1,3-diaryl-2-propen-1-one residue in molecules can be readily converted to various by-products by exposure to UV light. Natural chalcones can also be metabolized rapidly by drug metabolism enzymes, which causes a short half-life in vivo. For these reasons, numerous series’ of stable chalcone-like compounds have been obtained whose biological properties have been evaluated [132][134]. The most widely used method of obtaining chalcones is Claisen-Schmidt condensation [64][66]. During the reaction, the base (catalyst) attacks α-hydrogen of acetophenone and forms an enolate anion (carbanion). The carbanion attacks the carbonyl group of benzaldehyde and forms a β-hydroxycarbonyl intermediate, from which chalcone is obtained [133][134][135][136][137][138][139][135,136,137,138,139,140,141]. Synthetic chalcones can also be obtained by Suzuki reaction, Friedel-Crafts acylation, Wittig reaction and photo-Fries rearrangement of phenyl cinnamate [140][142]. Among synthetic chalcones, hybrid molecules with indole in the molecule are widely used in anticancer therapy. For example, Yan J et al. synthesized a series of 29 indole chalcones in order to evaluate their antiproliferative activity. The most active compound in the series, (E)-3-(6-Methoxy-1H-indol-3-yl)-2-methyl-1-(3,4,5-trimethoxyphenyl)propen-2-en-1-one (Compound 8), has IC50 values = 3–9 nM on six cancer cell lines. The new tubulin polymerization inhibitor binds to the colchicine binding site. Studies of cellular mechanisms show that derivative blocks the cell cycle in the G2/M phase and induces apoptosis consecutively with decreasing mitochondrial membrane potential. The indole compound has low cytotoxicity compared to normal human cells and has a similar potency to therapy-resistant cells [141][143]. Given the need to identify new anticancer therapies and the growing interest in identifying the biological properties of chalcones, reswearchers evaluated the antitumor mechanisms of these compounds.2.1.1. Anticancer Activity of Chalcones
The anticancer potential of chalcones is correlated with their ability to act on various molecular targets such as ABCG2 (ATP binding cassette subfamily G), tubulin, activated nuclear B cell growth (NF-κB), vascular endothelial growth factor (VEGF), tyrosine kinase receptor (EGFR), mesenchymal-epithelial transition factor (MET), 5-α reductase, ACP-reductase, histone deacetylase, p53, CDC25B (protein tyrosine phosphatase), retinoic acid receptors, estrogenic topoisomerase receptors and MDM2 [64][142][143][144][145][66,144,145,146,147]. In order to obtain chalcones with superior anticancer properties, three methods of modulation of natural chalcones were used: (1) modulation of substituents at the level of the two aromatic residues (aldehyde and acetophenone), (2) replacement of aromatic residues with heterocycles and (3) obtaining hybrid molecules by conjugating chalcones with other molecules with antitumor properties [64][66]. Since studies performed to highlight the anticancer activity of chalcones are numerous, and mechanisms by which they exert this property are multiple, reswearchers wanted to conduct a study on two important antitumor mechanisms of these compounds.2.1.2. ABCG2
ABCG2 (also called BCRP, breast cancer resistance protein) is a 655 amino acid protein that has a molecular weight of 72 kDa and consists of an N-terminal nucleotide-binding domain and a six-segment C-terminal transmembrane domain, as well as a hydrophobic and an extracellular loop between TM5 and TM6 [146][147][148,149]. Having a membrane localization, protein is part of the ATP-binding cassette (ABC) drug transporter family and has the ability to use the energy formed by hydrolysis of ATP [148][150]. It contains a transmembrane binding domain and a nucleotide-binding domain and is activated by homodimerization. Some high-performance 3D structures of ABCG2 protein binding to different substrates and inhibitors indicate the molecular mechanisms of ABCG2 substrate selection, binding and transport [149][151]. Many ABCG2 substrates are additional substrates for other ABC transporters, P-glycoprotein (ABCB1), so the net effect of the availability of a dual-substrate drug (ABCB1/ABCG2) is attributed to combined action of two transporters [150][152]. ABCG2 actively recognizes and transports distinct molecules, often hydrophobic, many of which have a polycyclic structure or a relatively flat shape [151][153]. BCRP is an efflux transporter with an important role in detoxification by removing endo- and xenobiotics from many cell types (e.g., uric acid, steroid metabolites), in modulating drug absorption and having an important role in various pharmacokinetic stages. Similarly, it determines resistance to anticancer therapy [152][153][154][155][156][157][154,155,156,157,158,159]. In addition, a transporter reduces the transfer of therapeutic substances to tumor cells. ABCG2 also has an important role in the protection of stem cells. It has been shown that the number of tumorigenic stem cells is directly proportional to the progression of cancers, these cells being involved in the initiation of tumorigenesis, tumor angiogenesis, metastases, drug resistance and recurrences. Stem cells are characterized by increased drug and chemotherapeutic resistance due to their expression in the efflux pump of ABCG2 [158][160]. It is located on the interface of the blood-brain barrier (efflux capacity of the protein protects the central nervous system from compounds with endogenous toxicity), placenta, liver, kidneys, colon and small intestine [159][160][161][162][163][164][165][161,162,163,164,165,166,167]. The transporter is involved in decreasing the oral bioavailability, tissue distribution and effectiveness of many therapeutic agents [166][168]. ABCG2 transporters are known to be responsible for limited exposure of the brain to anticancer drugs, the effectiveness of which reduces it, especially in case of brain metastases [167][169]. The clinical significance of BCRP expression is correlated with the response to therapy and the prognosis of gastrointestinal, lung, breast, head and neck tumors, ovarian, prostate, glioblastomas and leukemias and so forth [162][164]. BCRP has important functions in excluding antitumor drugs from various cancer cells, being a significant therapeutic barrier. Since it has subspecies polyspecificity and is present in many tissues, protein is an important factor in resistance to therapy [168][169][170][170,171,172]. In some tumor formations, ABCG2 is strongly overexpressed, which is correlated with unfavorable clinical outcomes for these tumors [171][173]. Because the carrier is crucial for the pharmacokinetics of certain compounds, the US Food and Drug Administration and European Medicines Agency have indicated that pharmacokinetic studies and drug–drug interactions have been performed for it [172][174]. Functional characterization indicates that ABCG2 carries a large number of different substances, including methotrexate, mitoxantrone, SN-38 and various tyrosine kinase modulators (e.g., imatinib, nilotinib), which act as both substrates and inhibitors of ABCG2. Multidrug resistance proteins 1 and 2, which are encoded by ABCG2 genes, play an important role in the excretion of tyrosine kinase inhibitors. In addition, ABCG2 recognizes a wide range of positively or negatively modified substances and is resistant to most topoisomerases I or II, such as topotecan or doxorubicin, which is the reason for therapeutic failure [173][174][175][176][177][175,176,177,178,179]. ABCG2 polymorphism explains the low accumulation of anticancer agents (doxorubicin, tyrosine kinase inhibitors, adriamycin, platinum compounds, sorafenib and mitoxantrone) in cells and their altered chemotherapeutic response [178][180]. Previous studies have indicated the architecture of BCRP and the structural basis of the inhibition of this protein by small molecules and antibodies [80][82]. The inhibition of these transporters has a favorable impact on anticancer therapy by co-administering them with chemotherapeutic agents. There is a growing need to identify selective inhibitors that optimize drug absorption and prevent possible side effects. Currently, there are a limited number of ABCG2 inhibitors, which are used as pharmacological tools in studies to highlight pathological/physiological role of this transporter, to cross the blood-brain barrier of these biologically active molecules and to reduce resistance to polytherapy [179][181].2.1.3. Chalcones with Activity on ABCG2
Licochalcone A (LCA, Compound 9, Figure 1), the most studied natural licochalcone, is present in roots of Glycyrrhiza inflata, has known pro-apoptotic and antiproliferative properties on different cell lines. Wu et al. determined the effect of LCA on multidrug-resistant ABCG2-overexpressing cancer cells. LCA has been shown to significantly alter the chemosensitivity of R482-HEK293 (ABCG2-transfected HEK293 human cells), S1-M1-80 (human colon cancer) and H460-MX20 (human non-small-cell lung cancer NSCLC) cells. For two known substrates of ABCG2 (mitoxantrone and topotecan), the activity was directly correlated with concentration [180][181][182,183].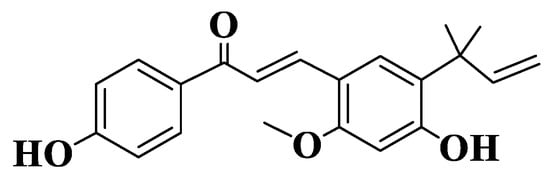
Figure 1. Structure of Licochalcone A.
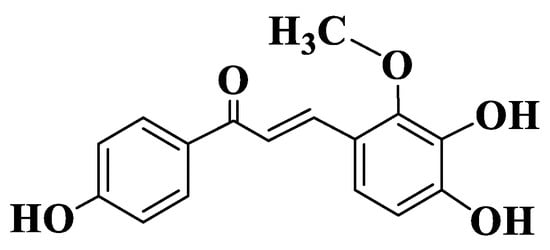
Figure 2. Structure of Licochalcone B.
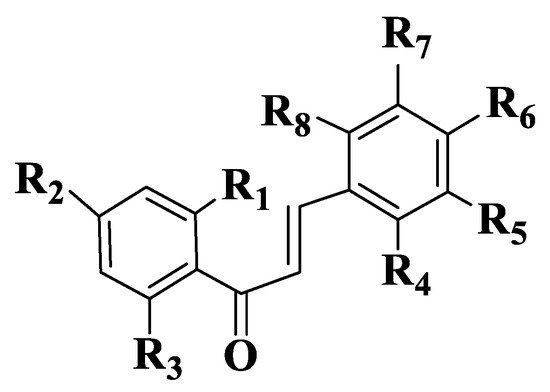
Figure 3. General structure of chalcones 14–47.
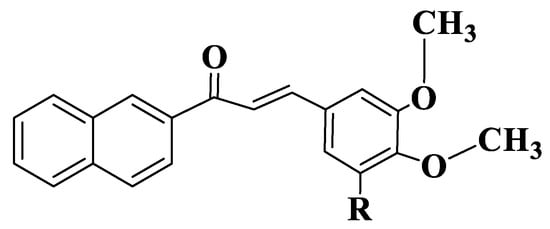
Figure 4. General structure of naphthochalcones (Compounds 58–61).
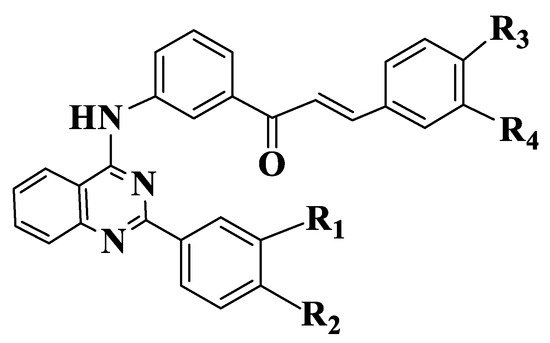
Figure 5. General structure of quinazoline chalcones (Compounds 66–71).
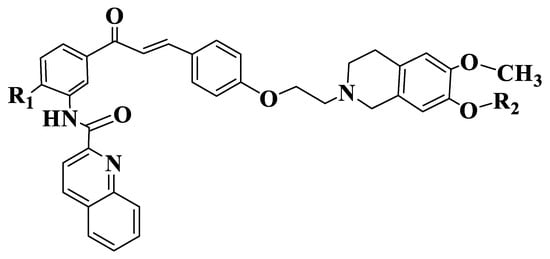
Figure 6. General structure of tariquidar-chalcones (Compounds 132–137).
2.1.4. Tubulin
Microtubules are cellular structures with a fundamental role in many essential biological processes, such as cytoskeletal architecture, intracellular transport, motility, chromosomal segregation and mitosis [90][190][191][192][193][194][195][92,192,193,194,195,196,197]. In the interface, microtubules are organized on cytoplasm in the form of a matrix-type network [196][198]. These are non-covalent mesoscopic polymers composed of α,β-tubulin protein heterodimers, which associate with and form protofilaments, which subsequently bind laterally and form microtubules. These dynamic structures are constantly elongated or shortened in all phases of cell cycle by adding or removing tubulin heterodimers from the ends of microtubules [197][198][199][200][199,200,201,202]. The assembly of protofilaments to microtubules is done spontaneously, with a number of protofilaments between 9 and 16, resulting in microtubules with different diameters [201][203]. It is known that the organization of microtubules changes significantly during the cell cycle. On the interface, microtubules form matrices in the cytoplasm and are relatively stable, and during mitosis, they form a bipolar mitotic spindle and become dynamic. When proliferative cells are exposed to microtubule inhibitors, bipolar spindle formation and the attachment of microtubules to kinetochores is inhibited, thus, activating the control point of division spindle assembly [202][204]. The stabilization of microtubules by acetylation is involved in cell migration [203][205]. Tubulin, in a depolymerized form, has the ability to influence cell physiology in various forms. For example, many microtubule-associated proteins have numerous sites of interaction with tubulin and microtubules. Thus, proteins containing the domain of overexpressed tumor genes use such multivalences to produce microtubule assembly. Mainly, tubulin, through its ability to influence the activity of proteins associated with microtubules, forms strong loops that connect the dynamics of microtubules to their basic matrix [204][206]. The dynamics of microtubules are strongly correlated with the regulation of cellular functions, such as intracellular transport and pathological processes [205][207]. This is essential for the optimal functioning of microtubules, in particular for the formation of dividing spindle in mitosis process. The disruption of this dynamic causes changes in cellular functions, influences the replication process and induces apoptosis. For these reasons, microtubules are one of the most studied targets of anticancer therapy. They actively participate in the formation of the centrosome, a formation characteristic of the G2/M phase of the cell cycle [206][207][208][209][210][208,209,210,211,212]. The unique feature of microtubule-binding agents, which is not present in other classes of anticancer agents, is their complexity and structural diversity, which determine many possibilities for optimization and modulation [211][213]. Compounds that interfere with cell division as a result of binding to α,β-dimers, oligomers or polymers have been extensively studied recently. Antimitotic agents, including different natural, synthetic or semi-synthetic products, have various chemical structures. Agents that interact with tubulin in the same binding region also belong to this category. Antimitotic agents include inhibitors of microtubule assembly (binds to Vinca domain or have sulfhydryl groups that alkylate tubulin) and microtubule-stabilizing agents (compounds that have a very high binding affinity, e.g., paclitaxel, docetaxyl, epothilone) [212][213][214][215][214,215,216,217]. Agents that target the field of colchicine (colchicine, podophyllotoxin, combrestatin A4) or those that bind to the domain of Vinca alkaloids (vincristine, vinblastine, vinorelbine) are defined as inhibitors of tubulin assembly as destabilizing agents [216][217][218][219][218,219,220,221]. Colchicine has a particular interest due to its antimitotic properties. The therapeutic potential of colchicine binding site has been investigated for its applications in chemotherapy. Colchicine binds strongly to the β subunit of tubulin. Cys241 residue forms hydrogen bonds with the trimethoxyphenyl residue of colchicine, and tubulin residues Thr179 and Val181 form hydrogen bonds with the troponelone residue of colchicine [216][220][218,222]. It is an increased need to identify new agents that bind to colchicine binding site. It is known that the intensity of the bond is dependent on the interaction of trimethoxyphenyl residue of colchicine with the binding site, and inhibition is achieved by interactions between oxygen atoms of the topolone network [221][223].2.1.5. Antitubulin Chalcones
Malik et al. evaluated the potential for the inhibition of microtubule polymerization by semisynthetic chalcones (Compound 138, (E)-1-(2-(allyloxy)-4-methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one, Figure 7). Assays were performed in vitro on HCT116 (human colorectal carcinoma) and MV-4-11 (human acute myeloid leukemia) cell lines. The derivative was observed to be a robust inducer of histone H3 to S10 phosphorylation after 8 h of treatment with 25 μM of the compound. The treatment of HCT116 or MV-4-11 cells with different concentrations of chalcone at 8 h resulted in a significant increase in histone H3 phosphorylation at S10, even at concentrations of 0.78 μM. Chalcone-induced cell cycle blockade was also confirmed by a FACS analysis of MV-4-11 cells. Through biochemical tests performed, it was observed that the derivative inhibits polymerization of microtubules depending on concentration (2–25 μM). Chalcone docking studies indicate its binding to a colchicine binding site, one of the aromatic residues being placed on trimethoxyphenyl test region of colchicine. The compound forms two hydrogen bonds with tubulin on Leu255 and Cys241 residues. The phenyl residue adjacent to the carbonyl group forms Π-σ interactions with Leu248, and the other aromatic residue interacts with residues Ala180 and Lys254 [222][224].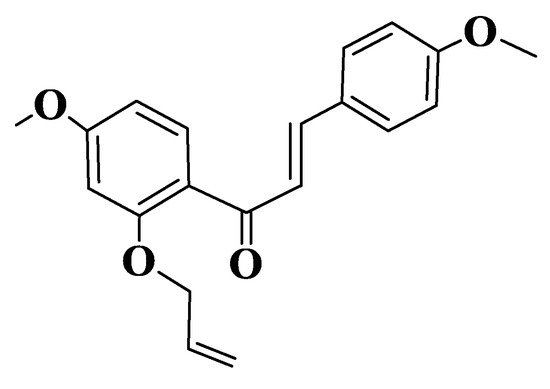
Figure 7. Structure of a semisynthetic chalcone (Compound 138).
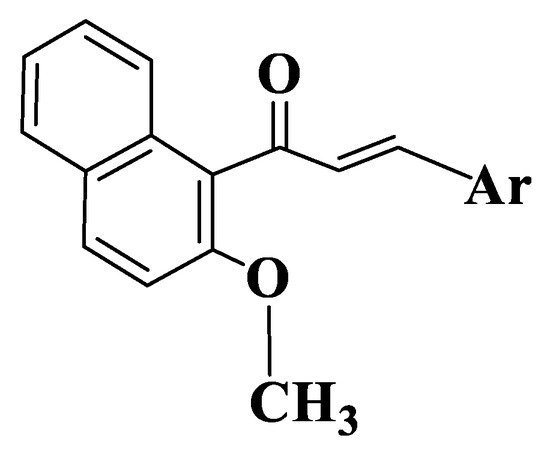
Figure 8. General structure of naphthalenchalcones (Compounds 142–161).
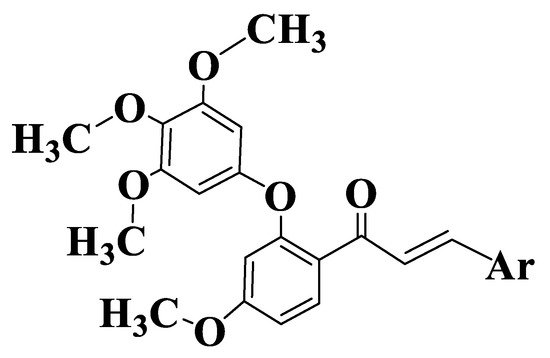
Figure 9. General structure of chalcones 162–177.
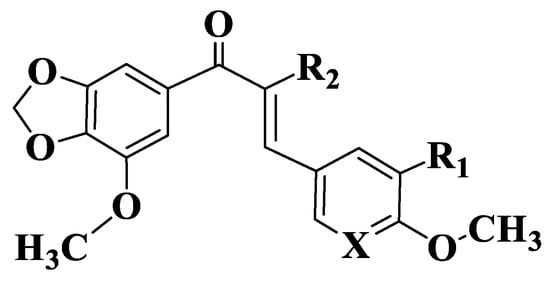
Figure 10. General structure of chalcones 179–187.
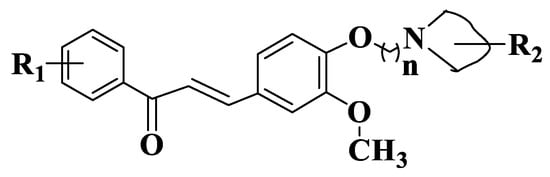
Figure 11. General structure of quinoline chalcones (Compounds 190–214).
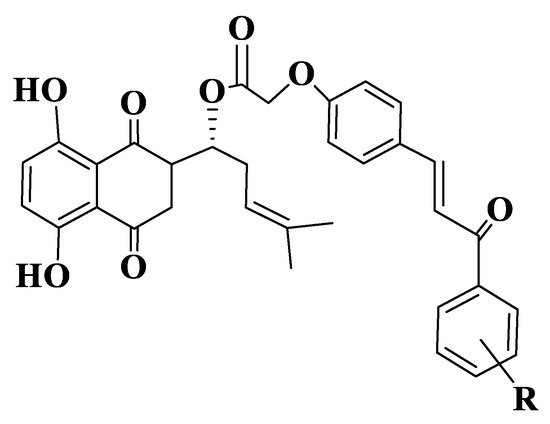
Figure 12. General structure of shikonium-derived chalcones (Compounds 215–232).