Global proteomic tools, such as liquid chromatography tandem mass spectrometry and matrix-assisted laser desorption/ionization time-of-flight, are used to profile the sperm proteins to identify the molecular pathways that are defective in infertile men. This article discusses the use of proteomic techniques to analyze the spermatozoa proteome. It also highlights the general steps involved in global proteomic approaches including bioinformatic analysis of the sperm proteomic data.
- proteomics
- bioinformatics
- sperm
1. Process and Techniques
The profiling of proteins extracted from a tissue or cell is termed as proteomics. Shotgun or bottom-up proteomics are common proteomic approaches to detect the proteins (>1000) in a very short period of time. Sperm proteins are detected using both conventional and advanced proteomic techniques. Two-dimensional (2D) gel electrophoresis is the most commonly used technique to separate sperm proteins based on the isoelectric point and molecular weight of peptides. The 2D-gel electrophoresis coupled with matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) technique was used to identify 98 distinct proteins in the human spermatozoa[1] [21]. A modified version of the 2D-gel electrophoresis technique, known as difference gel electrophoresis (DIGE), is used to identify differentially expressed proteins (DEPs) with a minimum error of <10%[2] [22]. Based on the intensity of the different staining dyes (Cy3 and Cy5), the expression of the DEPs aredetermined on the same gel using an automated image analysis software, such as Typhoon Trio Imager (GE Healthcare)[3] [23]. Advanced proteomic techniques include analysis of sperm proteins using MALDI-TOF and liquid chromatography-tandem mass spectrometry (LC-MS/MS). These instruments can detect the maximum number of proteins, even in samples that are of lesser concentration. Using the in-gel digestion based LC-MS/MS approach, Johnston et al. identified 1760 sperm proteins[4] [24]. Later on, several other studies also employed the LC-MS/MS-based proteomic profiling of spermatozoa in male infertility cases[5][6][7][8][9] [9,20,25–27]. In general, comparative protein analysis involves incorporation of stable isotopes for labeling the peptides. Such techniques include tandem mass tags (TMTs), isobaric tag for relative and absolute quantitation (iTRAQ) labelling, stable isotope labeling by amino acids in cell culture (SILAC) and isotope-coded affinity tag (ICAT). Among these techniques, iTRAQ labelling is widely used in sperm proteomics[9][10] [27,28]. Recently, an alternative label-free technique was developed to profile the proteins in sperm cells. Liu et al., used label-free proteomic approach to profile the proteins in the spermatozoa of asthenozoospermic obese men[11] [29]. Similarly, Moscatelli et al. also used the same approach to identify the protein involved in the bioenergetics pathways affecting sperm motility[12] [30].
2. Proteomic Analysis Using LC-MS/MS
Semen contains cellular (spermatozoa) and non-cellular (seminal plasma) components, the latter is enriched with proteins, thereby representing a target for seminal plasma proteomics and a putative source of markers[13][14] [31,32]. Other than spermatozoa, semen also contains round cells, which are of two types: Spermatogenic and non-spermatogenic round cells. Few studies have proposed that the use of sperm with round cells in the protein extraction process may contaminate sperm proteome. Separation of round cells and leukocytes are carried out using gradient centrifugation method and the pure fraction of the spermatozoa are used for proteomic analysis[15][16][17][18][19][20] [33–38]. Recently, Panner Selvam et al., investigated the interference of round cell proteins in the proteome of sperm and their effect on biological pathways associated with sperm function[21][22] [39,40]. The influence of non-spermatogenic round cell proteins was found to be negligible or insignificant when compared to that of the sperm proteome[21] [39]. Furthermore, the presence of these round cells and leukocyte proteins did not interfere in the molecular pathways associated with sperm function[22] [40]. However, while investigating the physiological functions of spermatozoa such as hyperactivation, capacitation and acrosome reaction, the use of pure fractions of normal sperm isolated using double gradient centrifugation or swim-up techniques has been reported[23][24] [41,42].
While processing sperm samples, the spermatozoa are first separated from semen or seminal fluid by centrifugation. The isolated sperm are washed 3 to 4 times with phosphate-buffered saline (PBS) to remove the remnants of seminal plasma. The sperm pellet is then mixed with lysis buffer (often radioimmunoprecipitation assay (RIPA) solution with SDS) and left overnight. This results in the complete lysis of spermatozoa[21] [39]. Sperm proteins are also extracted by sonicating the sperm cells suspended in an isotonic medium. Extracted sperm proteins are checked for their purity and concentration, and then subjected to electrophoresis (either one-dimensional or 2D-gel). Other methods, such as filter aided sample preparation (FASP) and in-solution, are also used to prepare samples for proteomic analysis. Samples subjected to FASP provide comparatively more proteome coverage, compared to samples processed using in-gel and in-solution digestion for mass spectrometry-based proteomics[25] [43]. Wang et al., used the FASP coupled MS/MS method to identify N-glycosylated proteins and glycosylation sites in human sperm[26] [44]. The protein extract can either be subjected to i) SDS PAGE and in-gel digestion, or ii) in-solution digestion, or iii) FASP (Figure 1).
MS detects the peptides and proteins using an unbiased approach[27] [45]. The proteins are indirectly inferred from the peptide with a very low false discovery rate (FDR), based on their mass/charge ratio (m/z). In general, the results of proteomics experiments contain 1–5% false positive protein identifications based on the inferred peptides using MS. To identify post-translational modification, such as acetylation, methylation and phosphorylation in the sperm proteome, enrichment protocols are recommended. In addition, LC-MS/MS is used to reduce the complexity of peptide mixture that was obtained after digestion of the proteins. Other techniques such as MALDI-TOF and SELDI-TOF (surface-enhanced laser desorption/ionization time-of-flight) are also successfully used to detect sperm proteins[28][29] [46,47].
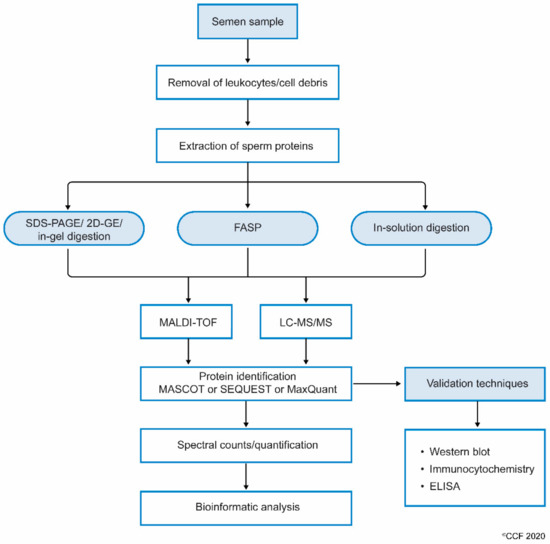
Figure 1.
Workflow involving the processing of semen samples for sperm proteomics.
Next, the complete scan of peptides detected by the MS are initially compared with the global database consisting of previously annotated and sequenced genes corresponding to respective proteins. At the least, masses of three peptide fragments should match with the corresponding homologous peptide masses in the protein database. Computational software such as SEQUEST, Mascot, MaxQuant and X!-Tandem, that operate using different algorithms, then display the complete list of proteins[30] [48]. In addition, the proteins are categorized as DEPs based on spectral counts (SC) and abundance of each protein. These DEPs are used in the downstream bioinformatic analysis to understand the role of proteins in molecular pathways[31] [49]. Gene ontology (GO) analysis provides additional information such as localization and functional annotation of proteins. Freely available bioinformatics tools such as STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) are used to display the interaction between proteins[32] [50]. In addition, commercially available sophisticated software such as Ingenuity Pathway Analysis (IPA) and Metacore™ are used to obtain a complete picture of the interactions between the proteins and the transcriptional factors regulating their expression[33] [51]. Furthermore, careful interpretation of the bioinformatics results is required for sperm, as these tools have been developed for somatic cells.