Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Gonzalo De Gonzalo and Version 2 by Catherine Yang.
The application of the so-called green solvents has emerged as a useful alternative to the classical organic solvents. These solvents must present some properties, such as a low vapor pressure and toxicity, high boiling point and biodegradability, and must be obtained from renewable sources.
- biobased solvents
- green chemistry
- biocatalysis
1. Biobased Solvents in Asymmetric Metal-Based Catalysis
Notably, a big portion of asymmetric synthesis includes chiral organometallic reagents, such as enantiomerically enriched organolithiums and Grignard reagents [1][52]. Organometallic reagents appear to show a better performance in solvents featuring ether functionalities due to their Lewis-base behavior, able to disaggregate organometallic species modulating their reactivity [2][53]. Unfortunately, the most employed solvents, such as THF, tend to react with highly basic carbanions, precluding the use of high temperatures in order to avoid undesired reactions, such as the α-cleavage of THF in the presence of lithium (e.g., the half-life in the presence of n-BuLi 10 min at 35 °C) [3][54]. The most serious drawback of ethereal solvents is the formation of peroxides (POs) under an oxygen atmosphere, limiting storage and handling safety, but also possible irritation, genotoxicity and mutagenicity during the exposure [4][55].
Ethereal solvents can dramatically affect the configurational stability of enantioenriched organometallic reagents, as showed for the enantioselective trapping of configurationally stable lithiated [5][6][56,57] and magnesiated nitriles [7][58], thus having an impact on the key characteristic for the synthetic utility of them in asymmetric synthesis. In the preparation of the enantioenriched cyclopropylnitrile Grignard reagent (S)-1, through the Mg/Br exchange of bromonitrile (S)-2 with i-PrMgCl, it exhibits a more configurational retention in Et2O < 2-MeTHF < THF (Figure 1), proving the role of ethereal solvent not only for the configurational stability but also for the stereochemical fidelity of the Mg/Br exchange, in accordance with Gawley and coworkers, as reported for the racemization rates of a lithiated Boc-pyrrolidine [8][59]. In fact, the coordinating ability of 2-MeTHF is intermediate between Et2O and THF, so that the rate of enantiomerization is faster than with Et2O but slower than with THF [9][60].
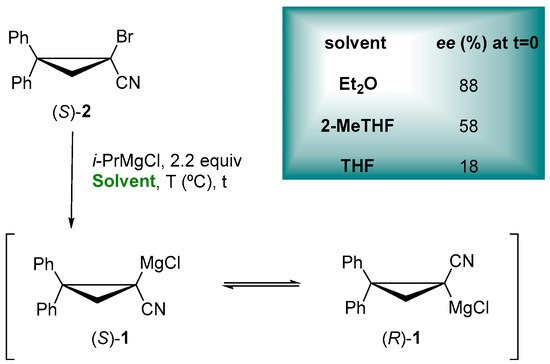
Figure 1.
Enantioenriched α-cyanocyclopropyl Grignard reagents.
The asymmetric synthesis of enantioenriched alcohols is representing a pivotal step in the synthesis of pharmaceutical molecules (Figure 2) [10][61]. The commonly employed methodologies involve the use of biocatalysts [11][62], transition metals [12][63], chiral hydrides [13][64] or homogenous asymmetric reduction based on oxazaborolidine [14][65].
Figure 2.
Pharmacologically active homochiral analogues.
Luisi’s group exploited the use of the Corey–Bakshi–Shibata (CBS) oxazaborolidine as a catalyst to control the enantioselectivity of the reduction of prochiral arylketones under homogeneous conditions in combination with the use of chip microreactors and 2-MeTHF as a greener solvent without employing any additives to develop a more sustainable process, as shown in Figure 3 [15][66]. In fact, the microreactors and continuous flow technologies have attracted the interest of the pharmaceutical and chemical industry due to the lower costs, improved reliability, safety and sustainability and the continuous processes, demonstrating, together with the solvent choice, their importance in the design of greener processes [16][67]. During the optimization phase, the screening of the solvents presented 2-MeTHF to provide a slightly better enantiomeric excess of the desired product in comparison with THF. Under the optimized reaction conditions, the targeted enantiopure alcohols were obtained in up to a 99% yield and 82% enantiomeric excess in only 10 min. In addition, the use of flow technology and the in-line monitoring and work up procedures helped to reduce the amount of the catalyst and optimize the process also in term of sustainability.
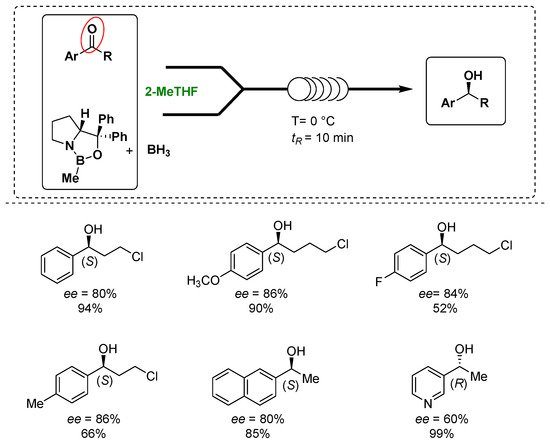
Figure 3.
Luisi’s asymmetric reduction of ketones to alcohols under microfluidic conditions in 2-MeTHF.
Starting from a series of aromatic and aliphatic trifluoromethylketones enroute to bench-stable trifluoromethyl N-tert-butanesulfinyl ketoimines (Figure 4, (3)), and subsequently reacting with the Reformatsky reagent formed in situ, Grellepois described the asymmetric synthesis of β-alkyl(aryl) β-trifluoromethyl β-amino acids (4) containing a quaternary stereocenter at the β position (Figure 4). The reaction was explored at room temperature in a variety of solvents (DME, Et2O, THF, 2-Me-THF, CH2Cl2, DMF or acetonitrile), showing a major stereoselectivity for the addition in 2-MeTHF. In addition, the use of this solvent was found beneficial in order to further decrease the temperature at 0 °C, thus having a better diastereoselectivity (88:12 instead of 86:14) and yield [17][68].
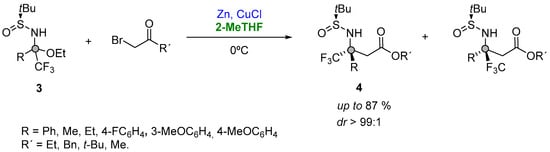
Figure 4.
Organozinc addition to a quaternary carbon center.
The introduction of a three-dimensional characteristic into a molecule, through the installation of a quaternary stereocenter, is representing an important tool to access various molecular scaffolds useful in drug discovery and design [18][2]. The evidence is, for example, the presence of nitrogen heterocycles in natural products or as useful building blocks in organic synthesis (Figure 5).
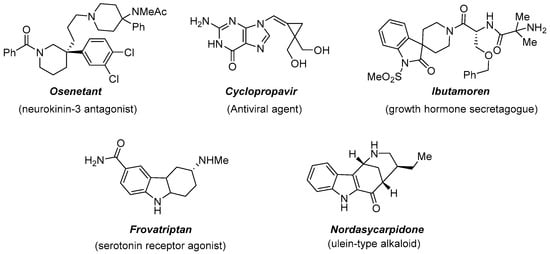
Figure 5.
Biologically active substances featuring a quaternary stereocenter.
Among asymmetric synthesis procedures, a particular advantage is represented by the use of transition metal catalysts, able to tune the interaction of a variety of ligands forming a number of possible diastereomeric combinations, and the interaction between the chiral metal complex and the achiral substrate represents the diastereotopic interaction leading to the asymmetric process [19][69]. In this context, Dyson and Jessop pointed out the importance of the interactions between a solvent, catalyst, substrates and products influencing both the speed and the selectivity of reactions [20][70]. Thus, solvents may tune the selectivity and efficiency of a given process and, in particular recent applications, show CPME [21][22][71,72] and 2-MeTHF [23][24][73,74] as useful green solvents in asymmetric catalysis.
Among all the examples in the enantioselective catalysis by metals, the ability of copper to coordinate a broad range of chiral ligands makes copper catalysis of particular interest [25][75]. Thus, the ability of copper to react with an electrophilic aminating reagent, such as 1,2-benzisoxazole, allowed the synthesis of enantioenriched β-amino acid derivatives, important building blocks for the synthesis of natural products and small molecule pharmaceuticals. Buchwald and coworkers reported an enantioselective CuH-catalyzed hydroamination protocol of α, β-unsaturated carbonyl compounds (cinnamate derivatives and tert-butyl cinnamate derivatives, Figure 6, (5)), with commercially available 1,2-benzisoxazole, in the presence of the ligand (S,S)-Ph-BPE. The reaction involves the in situ silylation of the carboxylic acid, the hydroamination to give the Schiff base, which upon hydrolysis releases the β-amino acid derivatives (6). The screening of the solvents indicated that CPME was an excellent solvent providing high yields and enantioselectivities (higher than 96% ee), as depicted in Figure 6 [26][76].
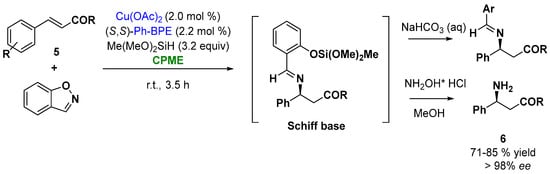
Figure 6.
Enantioselective Cu-catalyzed hydroamination of cinnamoyl derivatives.
Moreover, organometallic reagents are suitable for the enantioselective Cu-catalyzed conjugate addition to α,β-unsaturated carbonyl compounds [27][28][29][30][77,78,79,80]. Capable for this transformation, alkyl organoboranes or alkenyl organoboranes with the use of ferrocene-carbene ligands have also been presented. The catalytic activity of the ferrocene-carbene ligand L1 (Figure 7) was examined for the enantioselective addition of alkylborane to α,β-unsaturated ketones, using 15 mol% of CuCl with 15 mol% of chiral ligand in the presence of 30 mol% of tBuOK as a base. The target compound 7 was isolated in a 20% yield due to the decomposition of the ligand to vinylferrocene during the reaction and with 34% ee (Figure 7). To improve the reaction conditions from an environmental and health point of view, the “greener”, less hygroscopic and higher boiling solvent 2-MeTHF was employed. In fact, the use of THF, due to the high reaction temperature and time, resulted in the complete evaporation of the solvent. These prompt the use of different aromatic and etheric solvents (such as dioxane or toluene) which lead to the zero conversion into the product. On the other hand, the use of 2-MeTHF, due to the higher boiling point and its lower affinity to the water, resulted in the more efficient and suitable choice also in the case of the prolonged heating of the reaction mixture [31][81].
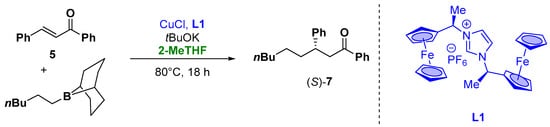
Figure 7.
Enantioselective Michael-type addition of organoboranes to chalcones.
The coordination of copper with the chiral ligand L2 (Figure 8) was also successfully employed in the case of copper-catalyzed asymmetric arylboronation of N-(o-iodoaryl) acrylamides (8) with bis(pinacolato)diboron (B2Pin2), leading to chiral oxindoles bearing BPin-containing all-carbon quaternary centers (9), as shown in Figure 8. The screening of the solvent indicated that a mixture of toluene/CPME was the optimal choice to achieve 96% ee and 62% yield [32][82].
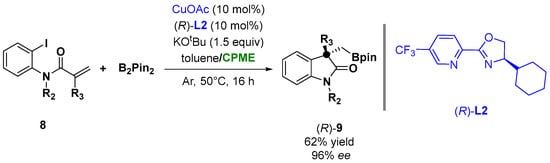
Figure 8.
Asymmetric α-borylmethyl oxindoles from
o
-iodoanilides.
The use of CPME appeared to also be productive, in a mixture 1:1 with MeCN, in the optimized synthesis of bortezomib ((10), Figure 9), an anti-cancer drug consisting of three structural units linked together by peptide bonds: pyrazinoyl, L-phenylalanyl and L-boroleucinyl. This compound was obtained via the stereoselective rhodium-catalyzed borylation of the N-adjacent C−H bond of the pro-boroleucine residue with bis(pinacolato)diboron (pinB-Bpin) in the presence of the triisopropylsilyloxy (TIPS)-modified BINOL-based mono-phosphite ligand at 80 °C for 36 h, furnishing bortezomib in a 53% yield and >98:2 d.r., after the transesterification of the pinacol ester with phenylboronic acid (Figure 9) [33][83].
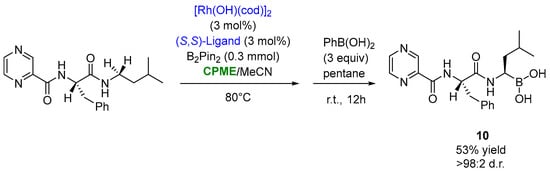
Figure 9.
Synthesis of bortezomib through asymmetric Rh-catalyzed borylation of aa leucine analogue.
Notably, in the asymmetric transfer hydrogenation (ATH) of α-alkoxy β-ketoesters (11) via dynamic kinetic resolution (DKR) involving a rhodium complex (N-pentafluorophenylsulfonyl-DPEN-based tethered Rh(III) complex, 2-MeTHF was selected as a greener solvent to develop an environmentally sustainable procedure [34][35][19,84]. ATH/DKR access the desired enantiomerically enriched syn-α-alkoxy β-hydroxyesters (12) in high yields (68–97%), high levels of diastereocontrol (95:5 to 99:1 d.r.) and excellent enantioselectivities (>99% ee) (Figure 10) [36][85].
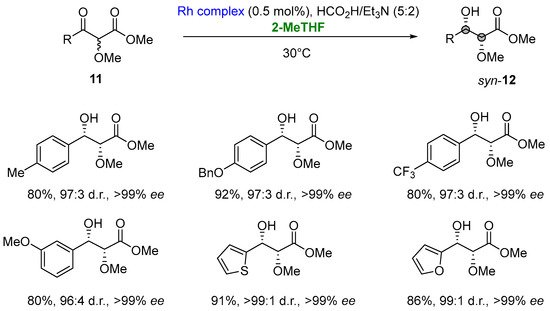
Figure 10.
Combined reductive-DKR en route to
syn
-α-alkoxy β-hydroxyesters.
The use of CPME and 2-MeTHF was also reported when carrying out asymmetric Nickel-catalyzed procedures as asymmetric alkylidenecyclopropanations and [2 + 2 + 2] cycloadditions to en route substituted pyridines, respectively. In the reductive Ni-catalyzed enantioselective alkylidene transfer from 1,1-dichloroalkenes (13) to olefins (14), the use of CPME as a solvent, in combination with the additive 1,3-dimethyl-2-imidazolidinone (DMI) and ligand L3, allowed the improving of the yields and enantioselectivity of the process at 0 °C, giving access to a broad range of alkylidenecyclopropanes (15), as indicated in Figure 11 [37][86].
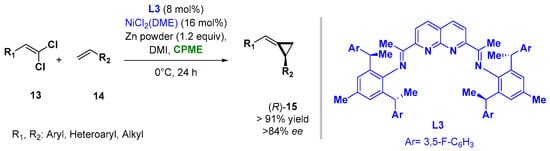
Figure 11.
Asymmetric Ni-catalyzed alkylidene-cyclopropanes from olefines and
gem
-dichloroalkenes.
The use of the bio-renewable 2-MeTHF provides a better yield and enantioselectivity in the case of a nickel-catalyzed asymmetric [2 + 2 + 2] cycloaddition in the presence of ligand (R)-L4 of the disubstituted malononitriles (16) and alkynes (17) to generate all-carbon quaternary center-containing substituted pyridines (18) under mild conditions. The use of zinc halide as an additive was crucial for the success of the process, and the better outcome of the reaction in 2-MeTHF appeared to be reasonable due to the weak coordination ability of this solvent to the transition metals and the high solubility of the zinc halides (Figure 12) [38][87].
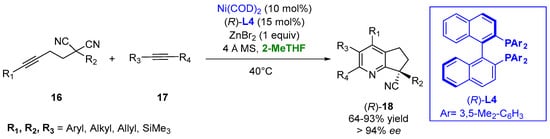
Figure 12.
Chiral pyridine derivatives from functionalized malononitriles and alkynes.
2. Biosolvents in Asymmetric Organocatalysis
The use of (relatively) small organic molecules as catalysts in organic reactions has experienced overwhelming interest in the last few years. The Nobel Prize in Chemistry in 2021 has confirmed the importance of this catalytic approach for the preparation of chiral compounds [39][40][41][42][88,89,90,91]. Organocatalytic methods offer some advantages over other catalytic approaches, such as the operational simplicity of the organocatalytic processes (normally tolerant to water and air), the stability and non-toxicity of the catalysts, the tolerance to different functional groups, the high diversity of the organic molecules available and the possibility of being modified, thus obtaining a wide set of compounds to be employed as organocatalysts in these reactions. Several approaches have also been performed to develop greener organocatalytic methodologies, thus including the application of green solvents.
In this context, the catalytic asymmetric alkylation of enolates was presented as one of the possible approaches for using biobased solvents. Thus, Maruoka and coworkers reported the asymmetric alkylation of 3-arylpiperidin-2-ones (19) under phase-transfer conditions in CPME to access a variety of δ-lactams (20) having a chiral quaternary carbon center at the α-position. The reaction of N-protected-3-phenylpiperidin-2-ones with alkyl and aryl bromides with the presence of the chiral quaternary ammonium salt (S)-I achieved the formation of the desired products in a high yield and enantioselectivity in short reaction time (Figure 13). The utility of the procedure was further demonstrated for the synthesis of osanetant, a neurokinin-3 antagonist [43][92].
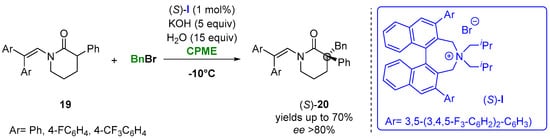
Figure 13.
Asymmetric α-benzylation of six-membered lactams in CPME.
The asymmetric allylic alkylation of the easily accessible Morita–Baylis–Hillman (MBH) carbonates of isatins (21) with a variety of enolizable cyclic carbonyl compounds catalyzed by the nucleophilic Lewis base 1,4-diazabicyclo [2.2.2]octane (DABCO, 10.0 mol%) leads to the synthesis of a novel class of spirooxindole-fused-dihydropyran scaffolds (22) in excellent yields and optimal diastereoselectivity, up to 99:1 (Figure 14a). The use of 2-MeTHF, an environmentally benign solvent, was chosen as the valuable and ecofriendly choice, ensuring the diasteroselectivity of the process [44][93].
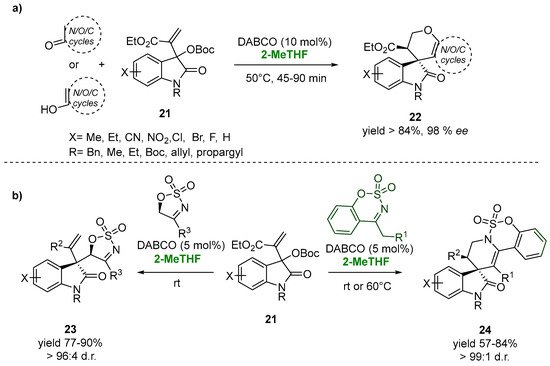
Figure 14.
3,3-disubstituted oxindoles via MBH-chemistry in 2-MeTHF (
a
,
b
).
This biobased solvent also proved its versatility in the case of the reaction of cyclic sulfamidate imines with MBH carbonates of isatins in the presence of a catalytic amount of DABCO for the synthesis of medicinally promising polycyclic spirooxindoles (23). The allylic substitution reaction of the MBH carbonates with five-membered cyclic sulfamidate imines at room temperature gave the desired products (23), showing, in 2-MeTHF, an excellent yield (87%) and diastereomeric ratio (96:4). In the case of the stereoselective synthesis of the 3,3-tetrahydropyridinyl spirooxindole scaffold (24), an increasing of the temperature to 60 °C appeared to be essential for the allylic alkylation-intramolecular aza Michael reaction sequence, also showing the versatility of the green solvent with respect to the variation of temperatures. After 6 h, the reactions resulted in 76–84% yields and excellent dr (99:1) (Figure 14b) [45][94].
CPME was efficiently employed in the construction of chiral polycyclic tetrahydrocarbazole (27) and chromane derivatives via an aminocatalytic enantioselective [4 + 2] Diels−Alder of β-indolyl α, β-unsaturated aldehydes (25) and α, β-unsaturated aldehydes (26) simultaneously activated by the aminocatalyst II in the presence of m-ClC6H4CO2H (m-ClBA), giving the best results in term of the yield and excellent stereoselectivities (in all cases, the ee was higher than 99%, d.r. >20:1) (Figure 15). Notably, also in the synthesis of the polycyclic chroman derivatives bearing four chiral centers, a remarkable stereoselectivity was obtained, carrying out the reaction in the same solvent [46][95].
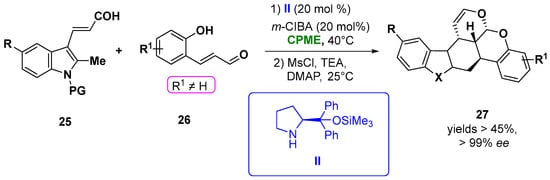
Figure 15.
Synthesis of chiral carbazole derivatives from unsaturated indolyl-aldehydes and enals.
Notably, in the asymmetric chiral phosphoric acid (CPA) catalyzed 1–6 addition of naphthols (28) to a set of para-quinone methides (p-QMs), prepared in situ from secondary p-hydroxybenzyl alcohols (6), CPME proved to be an effective green solution, replacing CHCl3 [47][96] and showing also a superiority in enantioselectivity. The methodology leads to the formation of tertiary stereocenters (29), overcoming the limitation of the use of stabilizing bulky substituents (e.g., t-Bu) at the α positions of the presynthesized p-QMs and the restriction in the construction of quaternary stereocenters from tertiary alcohols. The reaction catalyzed by the spirocyclic bis(indane)-derived chiral phosphoric acid III, and the use of 4 Å molecular sieves, which facilitate the generation of the p-QM intermediate removing the water generated during the process, gave access, at room temperature in 72 h, to a wide range of triarylmethanes in high yields and with excellent enantioselectivity (93% yield, 90% ee) (Figure 16) [48][97]. It is worth underlining that the phosphoric acid III serves as a bifunctional catalyst, activating both of the reaction partners via hydrogen bonds.
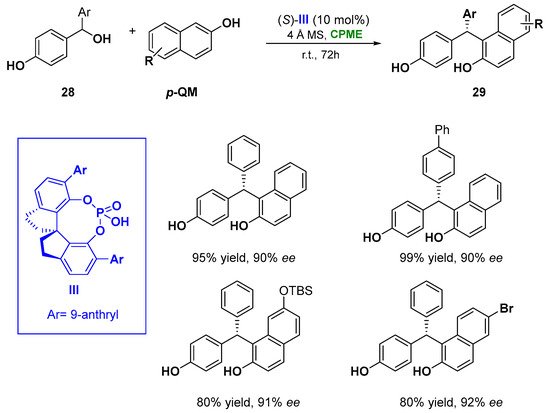
Figure 16.
Asymmetric CPA catalyzed of 1,2-diaryls from naphthols and benzylic alcohols.
Due to the major chemical stability of the α-CH bond of CPME, in comparison to other ethereal solvents, such as diisopropyl ether or THF, together with its relatively high boiling point (106 °C), CPME is a suitable alternative in radical reactions. In recent years, a variety of radical addition and reduction methodologies were reported with the use of this solvent, and in the case of photoredox asymmetric catalysis, it also appears to be a valid choice [49][98].
In point of fact, the enantioselective reduction of azaarene-based ketones (30) via visible light performed in CPME at −40 °C using 0.5 mol% of metal-free photosensitizer DPZ, 10 mol% 1,10-spirobiindane-7,70-diol (SPINOL)-based chiral phosphoric acid (CPA) catalyst IV and N-phenylpiperidine V (1.2 equiv. as additive) with the presence of 3 Å MS, furnished the corresponding reduced chiral alcohols (31) in 72 h with yields up to 93% and enantiomeric excesses (ee) higher than 91%, as indicated in Figure 17 [50][99].
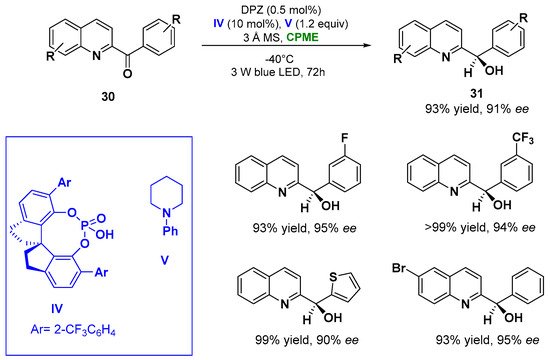
Figure 17. Photocatalytic enantioselective reduction of azaarene-based ketones in CPME employing a chiral phosphoric acid (IV) and N-phenylpiperidine (V).
The polystyrene-supported diamine VI, derived from L-tert-leucine, has been employed as a catalyst in the Robinson annulation to obtain the Wieland–Miescher (W-M) and the Hajos–Parrish (H-P) ketones (Figure 18) [51][100].
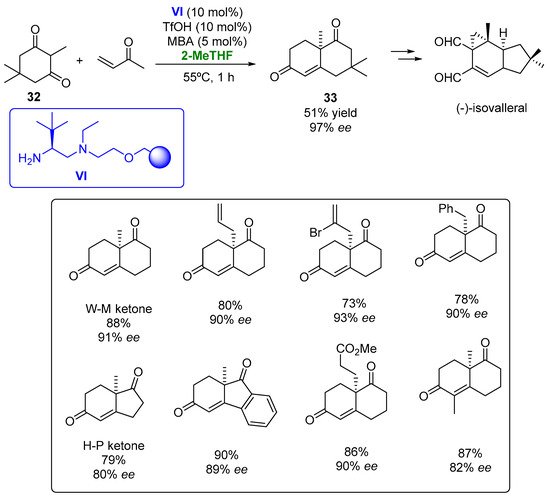
Figure 18.
Heterogeneous organocatalyzed preparation of bicyclic diketones employing 2-MeTHF as biobased solvent.
The heterogeneous catalysts were tested in the formation of the W-M ketone, starting from a triketone, with a complete conversion and 92% ee in the THF at room temperature being observed when using 10% mol triflic acid and 5 mol% m-nitrobenzoic acid (MBA) as the additive. When the reaction was performed in 2-MeTHF, a very similar result was achieved, so this greener alternative was established for carrying out these processes. When the reaction was carried out at 55 °C, a complete conversion was obtained after 1 h with an excellent optical purity (91% ee). The optimized conditions were employed for the formation of the W-M ketone, starting from diketone 32 and methyl vinyl ketone, achieving the desired compound after 1 h also with a complete conversion and high enantioselectivity. The substrate scope of this reaction was explored. Bicyclic ketones presenting a [4.4.0] structure and bearing different substituents at the 8a carbon atom were obtained in high optical purities (93% ee) after 1–2 h. The H-P ketone and some of its derivatives were also prepared by this methodology with excellent results. The enantioselective preparation of diketone 33, a valuable intermediate in the synthesis of furanether B or (+)-isovelleral was accomplished in the presence of catalyst VI and 2-MeTHF with a 51% yield and 97% ee, thus improving the optical purity of the previous methodologies described for the preparation of this compound. The robustness of the organocatalytic method was tested by recycling the catalyst in the preparation of the W-M ketone. High yields and optical purities were achieved after 10 cycles just by increasing the reaction times by 15 min from one reaction to the following one, thus indicating the robustness of the catalytic system.
In 2019, the 1,4-addition of α,α-dicyanoolefins (34) to chalcones (5), employing a bifunctional cinchona-derived organocatalyst (VII) in the presence of 2-MeTHF, was described [52][101]. The initial experiments were carried out in the reaction between the chalcone and α,α-dicyanoolefin in the presence of 9-amino-9-deoxyepiquinine (Figure 19, VII, 20 mol%) as the catalyst in THF, using trifle oroacetic acid (TFA, 20 mol%) as the cocatalyst. The chiral adduct (35) was recovered with a 56% yield and 97% ee after 4 days at room temperature. An evaluation of the reaction solvent led to the best results in the presence of 2-MeTHF, using both 15 or 20% v/v of the catalyst and cocatalyst, being possible to obtain the final adduct in 61% yield and 96% ee after 4 days. Once the reaction solvent was optimized, the procedure was extended to other substituted chalcones and α,α-dicyanoolefins, leading to a family of compounds with antiplasmodial properties in yields around 10–70% and optical purities between 71 and 98%.
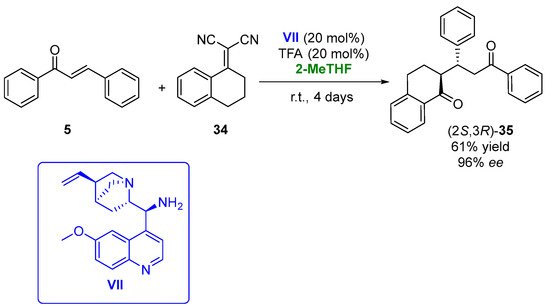
Figure 19.
Addition of α,α-dicyanoolefins to chalcones catalyzed by cinchona-based catalysts in 2-MeTHF.
Biobased solvents have been also applied in asymmetric hydrogen-bonding organocatalytic procedures. In the last few years, chiral squaramides have demonstrated their potential as this type of catalyst, being employed in several transformations with high regio- and/or enantioselectivities [53][102]. Thus, in 2018, the Michael/hemiketalization reaction of 4-hydroxycoumarines (36) with different types of enones (5) was carried out using squaramides as organocatalysts [54][103]. One of the processes analyzed was the addition of 4-hydroxycoumarin to benzylidenacetone yielding (R)-warfarin, a valuable anticoagulant (Figure 20a). After 5 days reaction at room temperature, 86% of the desired compound with 90% ee was recovered when employing a 1.4-dixoane/MeCN mixture and squaramide (R,R)-VIII (10 mol%) in the presence of 10 mol % of LiClO4 as the additive. Once the best catalyst was chosen, a solvent screening was performed, thus analyzing a set of deep eutectic solvents, ionic liquids and biobased solvents, including (-)-L-ethyl lactate, ethylene glycol and its dimethyl ether (monoglyme), PEG 600, CPME and 2-MeTHF. The use of PEG 600 afforded (R)-warfarin with an excellent optical purity (97% ee) but a modest yield, whereas the best results were obtained in 2-MeTHF (81% yield of enantiopure compound after 5 days). When squaramide (S,S)-VIII was employed as the catalyst, (S)-warfarine was obtained with good yield (84%) and a high optical purity (92% ee). The substrate scope of the Michael addition in 2-MeTHF was extended to several 4-hydroxycoumarins and benzylidenacetones. The use of different electronic groups in the hydroxycoumarin structure did not affect the optical purities of the final products, whereas this effect was observed for the benzylidenacetones. Those presenting electron-withdrawing groups led to high enantioselectivities, whereas those with electron-donating ones afforded the chiral adducts with low enantioselectivities (52% ee).
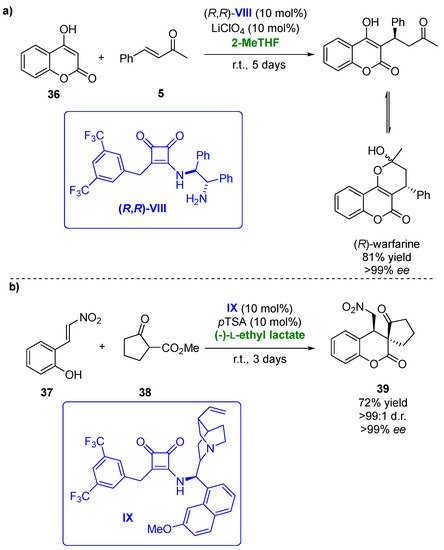
Figure 20. (a) Organocatalyzed synthesis of (R)-warfarin in presence of biobased solvents. (b) Michael addition and cyclation of 2-(2-nitrovinyl)phenols with β-ketoesters catalyzed by squaramide IX in biobased solvents.
Recently, a set of bifunctional squaramides have been tested as catalysts in the asymmetric sequential Michael addition and cyclization of a set of 2-(2-nitrovinyl)phenols [55][104]. These compounds reacted with cyclohexanone, yielding a lactol, which, after a reductive etherification, afforded hexahydro-1H-xanthenes. After catalyst screening, the best yields and optical purities were obtained when employing squaramide IX in the presence of (S)-proline as the cocatalyst and sodium acetate as the basic additive. Compound x was recovered with 83% yield, excellent diastereomeric purity (99:1) and 86% ee after 20 days when using DCM as the solvent. The Michael addition step of this sequential procedure was then tested in the presence of biobased solvents, including CPME, 2-MeTHF, (-)-L-ethyl lactate or ethylene glycol. The reactions were faster (8 days reaction time) than in DCM, but for all the solvents tested, lower yields and selectivities were achieved. The best choice for this process was ethylene glycol in the presence of (S)-proline and 4-nitrobenzoic acid as the additive, recovering the desired compound in a 61% yield with complete diastereoselectivity and 74% ee. The reaction between 2-(2-nitrovinyl)phenol (37) and cyclopentanone-2-carboxylate (38), a more reactive Michael donor, was also studied. When the process was carried out in DCM, employing IX in the absence of a cocatalyst and additive, final product 39 was obtained in a 66% yield with complete diastereoselectivity and 98% ee after 1 day (Figure 20b). The process was also analyzed in the presence of different biobased solvents. In this reaction, the use of (-)-L-ethyl lactate allowed to improve the catalytic procedure, as after 3 days, the enantiopure final compound was obtained with a 72% yield, thus demonstrating the advantages of employing this solvent.