Cellular senescence is a process resulting in stable cell cycle arrest, which restricts ability of cells to proliferate. It is considered that this state might be a response to chemotherapy, both genotoxic and oxidative stress, oncogenic activation, shortening of telomeres, irradiation or mitochondrial disorder. Senescence is generally considered as a process of tumor suppression, both by preventing cancer cells proliferation and inhibiting cancer progression. It can also be a key effector mechanism for many types of anticancer therapies such as chemotherapy and radiotherapy, both directly and through bioactive molecules released by senescent cells that can stimulate an immune response. Senescence is characterized by a senescence-associated secretory phenotype (SASP) that can have both beneficial and detrimental impact on cancer progression.
- senescence
- cancer
- therapy
1. Senescence Induction
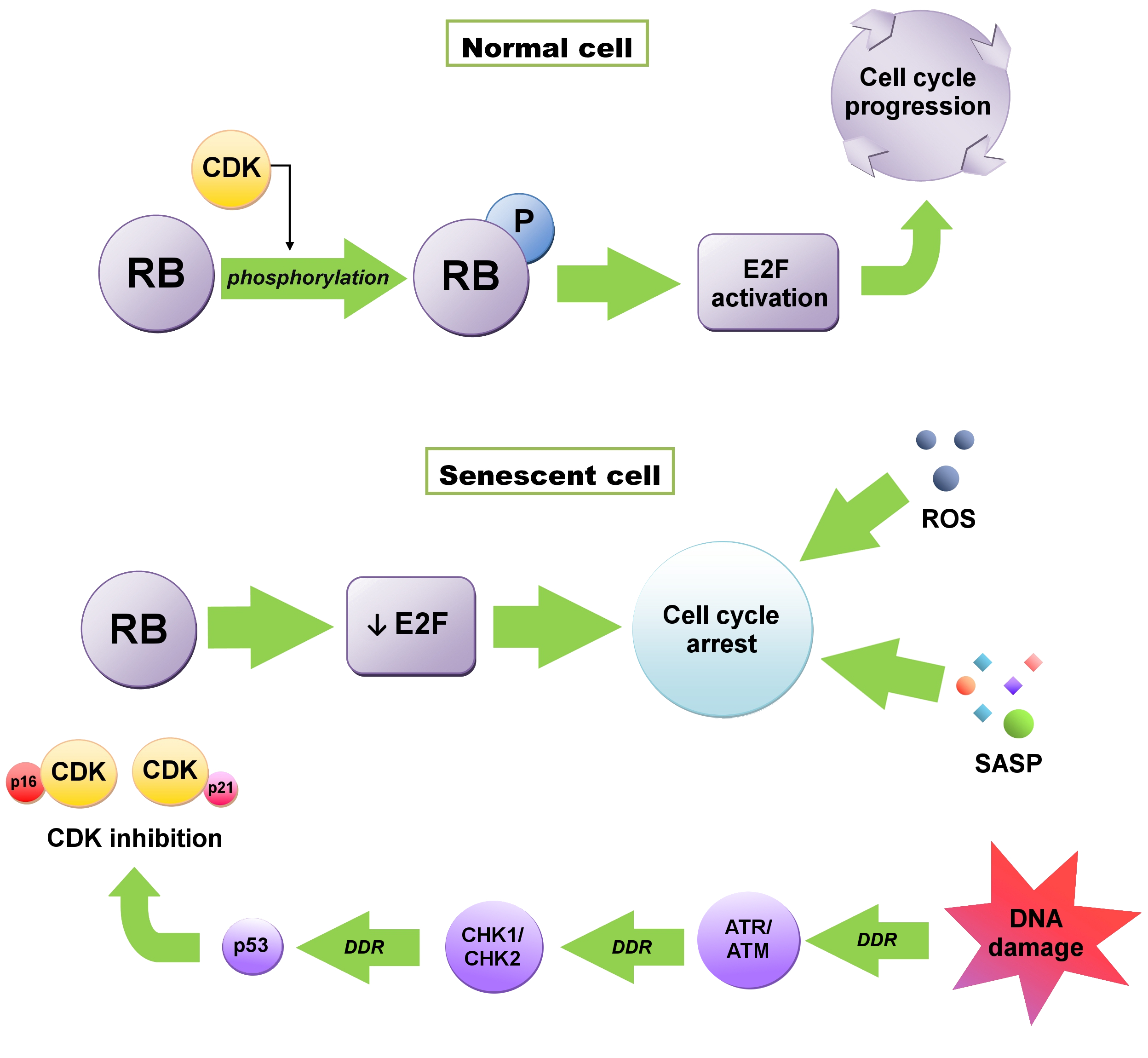
Induction of senescence is influenced by various factors, such as DNA damage, telomere shortening, hypoxia, nutrient deficiency, cell stress, oncogene activation, mitochondrial dysfunctions [1][3]. There is also a possibility of stimulating senescence through cellular metabolism, regulation of apoptosis, response to unfolded protein (UPR), and DNA damage response (DDR) [2][13]. Initiation of senescence is associated with the cell cycle, when a cell from the G1 phase, instead of going into the synthesis (S) phase, goes into the G0 phase, resulting in cell division inhibition. Activation of this checkpoint usually depends on DNA damage and telomere shortening [3][6]. Maintenance of this state, which is extremely important in anticancer therapy, is influenced by the cyclin-dependent kinase (CDK) inhibitors—P16INK4A (CDKN2A) and P21WAF1/CIP1 (CDKN1A) proteins—regulated by both the suppressor protein P53 and retinoblastoma proteins (RB) (Figure 1) [4][5][14,15]. RB1 protein, as well as P107 (RBL1) and P130 (RBL2), are phosphorylated by CDKs. The phosphorylation weakens their ability to repress E2F transcription factors, crucial to cell cycle progression [1][3]. In senescent cells, RB proteins are constantly activated due to the accumulation of the CDK4/6 inhibitor P16 and the CDK2 inhibitor P21. E2F transactivation is inhibited and consequently, cell cycle arrest occurs. This state is maintained by the SASP, heterochromatinization of E2F target genes and reactive oxygen species (ROS) and cannot be reversed by P53 or RB inactivation [1][6][3,16].21. Replicative Senescence
According to the Hayflick theory, cells’ ability to replicate is limited [7][17]. After reaching the limit, they become senescent cells—alive and metabolically active, but unable to divide. The main factor causing replicative senescence is the shortening of telomeres and the lack of telomerase responsible for their extension [8][9][18,19]. Telomeres are located at the ends of the chromosomes and are tandem repeats of TTAGGG nucleotides, stabilized by the Shelterin protein complex [8][9][18,19]. The majority of cells (except for stem and cancer cells) do not express telomerase responsible for maintaining telomere length [1][3]. With each replication, the length of telomeres decreases [3][6]. Eventually, the free end of the chromosome is exposed, which is perceived as a double-strand DNA break (DSB). DSBs activate the DDR [10][4].32. Stress–Induced Senescence
As mentioned before, senescence is associated with DDR. DNA damage caused by the above-described telomere shortening, induction of an oncogene, as well as damage due to the oxidative stress, external factors, and chemotherapeutic agents result in a cellular response [3][6]. A strong inducer of DDR is also the sugar-phosphate DNA backbone damage—caused by, among others, ionizing radiation (IR) or topoisomerase inhibitors—which may cause DSBs [11][20]. In case of constant DNA damage signaling, DSB contributes to the enhanced secretion of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and onset of inflammation [12][21]. Diminished selective autophagy of GATA binding protein 4 (GATA4)—transcriptional factor—also contributes to the induction of DDR-dependent senescence and inflammation. During senescence—due to the action of ataxia telangiectasia mutated kinase (ATM) and ataxia telangiectasia and Rad3-related kinase (ATR)—activation of P16INK4A and P53 pathways occurs. As a result, p62-dependent autophagic GATA4 degradation is inhibited. Activation of nuclear factor kappa B (NF-κB) occurs, which leads to the SASP initiation [13][22]. This process seems to be independent of P16INK4A and P53 [11][20]. Furthermore, telomere related factors such as protection of telomeres 1 (POT1) and telomeric repeat-binding factor 2 (TRF2) may impair the activity of DDR via inhibiting ATM and ATR kinases involved in the cell response to DNA damage [14][15][23,24]. Eradication of TRF2 or POT1 from telomeric DNA leads to DDR [16][25].