Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Júlia Sala and Version 2 by Conner Chen.
Tauopathies are a group of neurodegenerative diseases characterized by the hyperphosphorylation and deposition of tau proteins in the brain. In Alzheimer’s disease, and other related tauopathies, the pattern of tau deposition follows a stereotypical progression between anatomically connected brain regions. Increasing evidence suggests that tau behaves in a “prion-like” manner, and that seeding and spreading of pathological tau drive progressive neurodegeneration.
- tauopathies
- neurodegeneration
- seeding
1. Introduction
Tauopathies are a group of over 25 different neurodegenerative diseases (NDs), including, among others, Alzheimer’s disease (AD), primary age-related tauopathy (PART), progressive supranuclear palsy (PSP), chronic traumatic encephalopathy (CTE), Pick’s disease (PiD), corticobasal degeneration (CBD), or globular glial tauopathy (GGT) [1][2][3]. They are all characterized by the progressive accumulation of the microtubule (MT)-associated protein tau (MAPT) into insoluble aggregates. In parallel to tau aggregation, patients suffer from neuron loss and increased cognitive decline. Although considerable efforts have been made to overcome the pathophysiological aspects of tauopathies, there are currently no effective therapies for affected individuals.
Tau is an MT-binding protein that plays a fundamental role in modulating neuronal MTs dynamics in the axons of neurons [4]. Moreover, it is also present in oligodendrocytes and astrocytes, although with lower expression levels [5][6]. Importantly, under pathological conditions, tau dissociates from MTs and aggregates into highly insoluble, abnormally phosphorylated forms [7][8]. Indeed, tau aggregates are the main histopathological hallmark shared by all tauopathies. However, the primary cell types affected (neurons or glial cells), the regional distribution of the aggregates, and the morphology of these aggregates vary by disease [9] (e.g., storm-like, diffuse plaques, etc.). In AD, for example, tau aggregates form paired helical filaments (PHFs) and straight filaments, which contribute to the formation of neurofibrillary tangles (NFTs) [10]. In addition, tau aggregates from different tauopathies are biochemically distinct, as they differ in their extent of protease resistance, resulting in unique Western blot banding patterns [11][12], or different structures revealed in recent studies using cryo-electron microscopy (cryo-EM) [13].
To date, the relationship between tau aggregates and pathogenicity remains unclear. Thus, tau aggregates could either be a co-occurrence of another unidentified underlying disease or a direct cause of neurodegeneration. The latter is supported by the fact that mutations in the MAPT gene on chromosome 17 [14] cause inherited frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [12][15], implying a causal link between tau malfunction and neurodegeneration. In the case of sporadic human tauopathies, the gradual appearance of tau aggregates positively correlates with neurodegeneration [16]. Moreover, the presence of insoluble tau aggregates during the course of the disease is not limited to specific brain areas but appears to spread throughout the brain. For example, in patients with AD, histopathological tau lesions progress in a predictable, stereotyped, and hierarchical pattern along neuroanatomically connected brain regions (which has been described in neuropathological studies [17][18] and more recently confirmed via positron emission tomography (PET) [19]). However, the specific molecular and cellular mechanisms underlying the progression of tau pathology are still unclear. Since 2009 [20][21], there has been increasing evidence pointing to the belief that tau can spread between cells in a “prion-like” manner. The “prion-like” hypothesis states that seeded aggregation and cell-to-cell spread of pathological tau are two fundamental pathological aspects of human tauopathies (see [22] as a recent example).
2. Tau Structure and Processing
In the healthy adult human brain, there are six tau isoforms ranging from 352 to 441 amino acids in length. These isoforms are the products of alternative mRNA splicing of exons 2, 3, and 10 [23][24]. Exon 10 inclusion results in the production of tau isoforms with four repeats (4R), whereas its exclusion produces isoforms with three repeats (3R). The adult human brain expresses both 3R and 4R tau at equivalent ratios [25]. While tau aggregates in the brains of AD patients maintain this proportion, other tauopathies favor one isoform over the other. Thus, human tauopathies can be classified as 3R, 4R, or 3R/4R (e.g., PiD, GGT, and AD, respectively), determined by the isoforms present in the aggregates [26]. Tau is a highly soluble and intrinsically disordered protein that lacks a well-defined secondary structure [27]. Therefore, depending on interactions with different binding partners, it can adopt various conformations [25]. Despite this, tau can be divided into four functional domains: (a) the N-terminal domain, also known as the projection domain; (b) the proline-rich mid-domain, with different phosphorylation residues; (c) the MT-binding domain (MTBD) or repeat domain (RD), which consists of three (R1, R3, and R4) or four (R1-R4) MTs-binding repeats, due to the absence or presence of exon 10, respectively, and (d) the C-terminal region (Figure 1).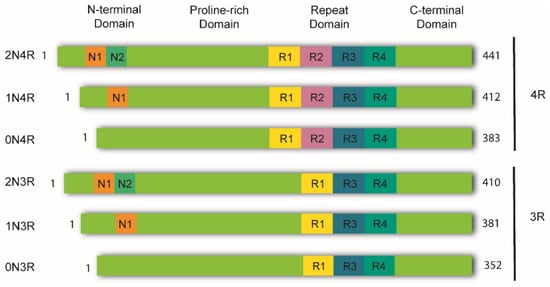
Figure 1. Schematic representation of tau isoforms. In the adult human brain, tau is found as six major isoforms (352-441 amino acids) resulting from alternative mRNA splicing. The N-terminal domain consists of either 0, 1, or 2 inserts encoded by exons 2 and 3 (0N, 1N, or 2N). The proline-rich domain is followed by the repeat domain (RD) also known as the microtubule-binding domain (MTBD). Here, inclusion of exon 10 produces tau isoforms with four repeats (4R), whereas its exclusion produces isoforms with three repeats (3R). The RD is followed by the C-terminal domain.
3. The “Prion-like” Nature of Tau and Its Strains
As previously mentioned, the progressive accumulation of tau results in dysfunction in brain regions affected by tau pathology [34]. In the early 1990s, Braak and Braak conducted a cross-sectional neuropathological study of the brains of AD-deceased patients [17] and established that the development of tau pathology occurs in a hierarchical fashion. Their work showed that the progression of NFTs does not occur randomly in the brain. Instead, tau lesions first accumulate in the locus coeruleus, from where they tend to spread to the transentorhinal region of the temporal lobe, followed by the allocortex and neocortex (first in associational areas and later in the primary sensory cortex and the primary motor cortex) [35]. Interestingly, some cellular types appear to be more vulnerable to tau aggregation than other cells [34][36][37][38]. Taken together, these findings could be interpreted in the context of pathological tau spreading in the brain through trans-cellular propagation. The idea that tau spreads through cell-to-cell transmission is commonly referred to as the “prion-like” hypothesis. Since 2009, there has been an increasing body of evidence supporting the notion that tauopathies and prion diseases (PrDs) share some common pathological mechanisms [3][20][21][39]. Indeed, not only tauopathies but also other NDs with amyloid deposition—also called “neural proteinopathies” (e.g., synucleinopathies)—are now referred to as “prion-like” diseases because they all seem to have replication and propagation processes akin to those observed in PrDs. According to the “protein-only” hypothesis, PrDs are a group of NDs caused exclusively by an infectious protein or prion known as the proteinase K-resistant prion protein (PrPSc) [39], which is the pathological form of the cellular prion protein (PrPC) [40]. Importantly, PrPSc forms insoluble amyloid aggregates that spread across brain regions by cell-to-cell transmission. Nonetheless, “prion-like” diseases are not currently considered bona fide prions [41][42][43] because, unlike prions, there is no conclusive evidence of interindividual transmissibility (see [44]). Therefore, such terminology has been used to specify these proteins that have similar replication and propagation processes as PrDs but are not infectious. Nevertheless, the fact that PrDs and “prion-like” diseases may share some pathological mechanisms could have significant implications for the development of novel treatments. The “prion-like” hypothesis proposes that, initially, only a small number of neurons initiate the process of tau aggregation. In these cells, tau misfolds and recruits its endogenous counterparts, templating the misfolding state via a process similar to the growth of crystals [45], known as nucleation-dependent polymerization. Replication of the misfolding state is a defining feature of “prion-like” behavior referred to as “seeding,” and the term “seed” describes the minimal unit needed to template the misfolding state of tau. Once the seed is formed, it begins the process of self-seeded fibrillization, in which tau monomers are progressively recruited and added to the growing fibril. Mature tau aggregates (or fibrils) exhibit amyloid properties (e.g., cross-β structure, Thioflavin-positive staining, resistance to detergents). Large tau aggregates can then be fragmented, creating new fibrils or seeds, thereby amplifying the pathology. At this point, competent seeds can be released and propagated to adjacent or synaptically connected healthy cells [33]. The stable propagation of tau aggregates is another characteristic of the prion paradigm behavior and is termed “spreading.” Both seeding and trans-cellular spreading of proteopathic tau seeds have been proposed as the pathological mechanism to explain the stereotyped progression of neurodegenerative tauopathies. Therefore, since tau is an intracellular protein, cell-to-cell spreading implies, in a broad sense, a four-step process (Figure 2): (1) one cell harboring intracellular tau aggregates (termed donor neuron), (2) release of competent tau seeds from the donor cell into the extracellular space, (3) internalization by a neighboring healthy cell (termed receptor neuron or glia), and (4) once inside, recruitment of their endogenous counterparts initiating further seeding.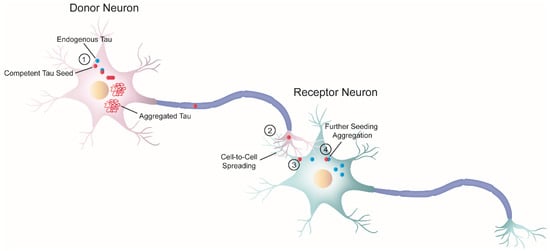
Figure 2. Schematic diagram of cell-to-cell progression of tau pathology. j The formation of tau aggregates begins in a donor neuron (pink) when a misfolded seed-competent tau (red) templates its misfolded state to its endogenous monomeric counterpart (blue), through a process known as seeding. Ultimately, the seeding process produces tau aggregates with amyloid properties. In parallel, tau seeds travel along the axon to the synaptic terminal of the donor neuron. k Once there, tau is released or transferred from the donor neuron to the receptor neuron (greenish-blue). Although not depicted here, glial cells could also internalize misfolded tau seeds. l Next, the receptor neuron internalizes seeded-competent tau. This diagram depicts only one of the several proposed mechanisms related to trans-cellular spreading, in which free tau seeds are released from the axon terminal and are internalized by the receptor neuron through direct membrane fusion. However, numerous studies have proposed a variety of cellular pathways involved in the progression of pathological tau, as reviewed by in steps k and l [46][47][48]. The exact nature of the pathological tau involved in the cell-to-cell transfer process is also unknown, and different groups have proposed a variety of candidates [49][50][51]. m Inside the receptor neuron, pathogenic tau can recruit endogenous cellular tau and seed further tau aggregation. Overall, this process ensures the progression of the pathology.
References
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672.
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225.
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 2014, 82, 1271–1288.
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378.
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373.
- Shin, R.W.; Iwaki, T.; Kitamoto, T.; Tateishi, J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab. Investig. 1991, 64, 693–702.
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566.
- de Garcini, E.M.; Serrano, L.; Avila, J. Self assembly of microtubule associated protein tau into filaments resembling those found in alzheimer disease. Biochem. Biophys. Res. Commun. 1986, 141, 790–796.
- Götz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 239–261.
- Hernández, F.; Ferrer, I.; Pérez, M.; Zabala, J.C.; del Rio, J.A.; Avila, J. Tau Aggregation. Neuroscience 2022.
- Höglinger, G.; Respondek, G.; Kovacs, G. New classification of tauopathies. Rev. Neurol. 2018, 174, 664–668.
- Kovacs, G.G. Tauopathies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 355–368.
- Chung, D.-E.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 2021, 16, 57.
- Dugger, B.N.; Whiteside, C.M.; Maarouf, C.L.; Walker, D.G.; Beach, T.G.; Sue, L.I.; Garcia, A.; Dunckley, T.; Meechoovet, B.; Reiman, E.M.; et al. The Presence of Select Tau Species in Human Peripheral Tissues and Their Relation to Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 54, 1249.
- Goedert, M.; Jakes, R. Mutations causing neurodegenerative tauopathies. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2005, 1739, 240–250.
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639.
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259.
- Braak, H.; Braak, E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278; discussion 278–284.
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D.; et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 2016, 89, 971–982.
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the Inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852.
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913.
- Franzmeier, N.; Brendel, M.; Beyer, L.; Slemann, L.; Kovacs, G.G.; Arzberger, T.; Kurz, C.; Respondek, G.; Lukic, M.J.; Biel, D.; et al. Tau deposition patterns are associated with functional connectivity in primary tauopathies. Nat. Commun. 2022, 13, 1362.
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526.
- Avila, J. Tau protein, the main component of paired helical filaments. J. Alzheimer’s Dis. 2006, 9, 171–175.
- Avila, J.; Jiménez, J.S.; Sayas, C.L.; Bolós, M.; Zabala, J.C.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262.
- Kovacs, G.G.; Ghetti, B.; Goedert, M. Classification of diseases with accumulation of Tau protein. Neuropathol. Appl. Neurobiol. 2022, 48, e12792.
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297.
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. 2004, 112, 813–838.
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471.
- Medina, M.; Hernández, F.; Avila, J. New Features about Tau Function and Dysfunction. Biomolecules 2016, 6, 21.
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68, 1067–1081.
- Avila, J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006, 580, 2922–2927.
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Alavi Naini, S.M.; Mandelkow, E.-M.; Mandelkow, E.; Buee, L.; Goedert, M.; et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017, 5, 99.
- Bancher, C.; Braak, H.; Fischer, P.; Jellinger, K.A. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci. Lett. 1993, 162, 179–182.
- Braak, H.; Zetterberg, H.; Del Tredici, K.; Blennow, K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-β changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. 2013, 126, 631–641.
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358.
- Fu, H.; Possenti, A.; Freer, R.; Nakano, Y.; Villegas, N.C.H.; Tang, M.; Cauhy, P.V.M.; Lassus, B.A.; Chen, S.; Fowler, S.L.; et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci. 2018, 22, 47–56.
- Braak, H.; Rüb, U.; Schultz, C.; Del Tredici, K. Vulnerability of cortical neurons to Alzheimer’s and Parkinson’s diseases. J. Alzheimer’s Dis. 2006, 9, 35–44.
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383.
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144.
- del Río, J.A.; Ferrer, I.; Gavín, R. Role of cellular prion protein in interneuronal amyloid transmission. Prog. Neurobiol. 2018, 165–167, 87–102.
- Vascellari, S.; Manzin, A. Parkinson’s Disease: A Prionopathy? Int. J. Mol. Sci. 2021, 22, 8022.
- Goedert, M. Tau proteinopathies and the prion concept. Prog. Mol. Biol. Transl. Sci. 2020, 175, 239–259.
- Coca, J.R.; Eraña, H.; Castilla, J. Biosemiotics comprehension of PrP code and prion disease. Biosystems 2021, 210, 104542.
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058.
- Brunello, C.A.; Merezhko, M.; Uronen-Mattila, R.-L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell. Mol. Life Sci. 2020, 77, 1721–1744.
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 707268.
- De La-Rocque, S.; Moretto, E.; Butnaru, I.; Schiavo, G. Knockin’ on heaven’s door: Molecular mechanisms of neuronal tau uptake. J. Neurochem. 2020, 156, 563–588.
- Mirbaha, H.; Holmes, B.; Sanders, D.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903.
- Sharma, A.M.; Thomas, T.L.; Woodard, D.R.; Kashmer, O.M.; Diamond, M.I. Tau monomer encodes strains. eLife 2018, 7, e37813.
- Kim, D.; Lim, S.; Haque, M.; Ryoo, N.; Hong, H.S.; Rhim, H.; Lee, D.-E.; Chang, Y.-T.; Lee, J.-S.; Cheong, E.; et al. Identification of disulfide cross-linked tau dimer responsible for tau propagation. Sci. Rep. 2015, 5, 15231.
- Vaquer-Alicea, J.; Diamond, M.I.; Joachimiak, L.A. Tau strains shape disease. Acta Neuropathol. 2021, 142, 57–71.
- Taniguchi-Watanabe, S.; Arai, T.; Kametani, F.; Nonaka, T.; Masuda-Suzukake, M.; Tarutani, A.; Murayama, S.; Saito, Y.; Arima, K.; Yoshida, M.; et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2015, 131, 267–280.
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540.
- Ferrer, I.; Andrés-Benito, P.; Zelaya, M.V.; Aguirre, M.E.E.; Carmona, M.; Ausín, K.; Lachén-Montes, M.; Fernández-Irigoyen, J.; Santamaría, E.; del Rio, J.A. Familial globular glial tauopathy linked to MAPT mutations: Molecular neuropathology and seeding capacity of a prototypical mixed neuronal and glial tauopathy. Acta Neuropathol. 2020, 139, 735–771.
- Goedert, M. Cryo-EM structures of τ filaments from human brain. Essays Biochem. 2021, 65, 949–959.
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140.
- Han, Z.Z.; Kang, S.G.; Arce, L.; Westaway, D. Prion-like strain effects in tauopathies. Cell Tissue Res. 2022.
More