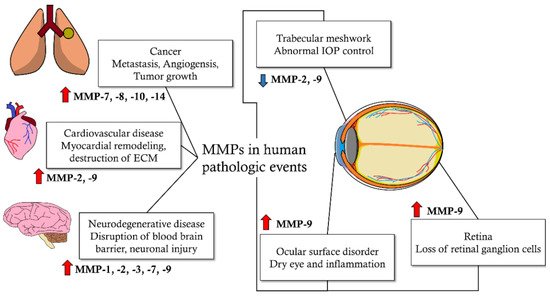
Matrix metalloproteinases (MMPs) are enzymes that decompose extracellular matrix (ECM) proteins. MMPs are thought to play important roles in cellular processes, such as cell proliferation, differentiation, angiogenesis, migration, apoptosis, and host defense. MMPs are distributed in almost all intraocular tissues and are involved in physiological and pathological mechanisms of the eye. MMPs are also associated with glaucoma, a progressive neurodegenerative disease of the eyes. MMP activity affects intraocular pressure control and apoptosis of retinal ganglion cells, which are the pathological mechanisms of glaucoma. It also affects the risk of glaucoma development based on genetic pleomorphism. In addition, MMPs may affect the treatment outcomes of glaucoma, including the success rate of surgical treatment and side effects on the ocular surface due to glaucoma medications. Glaucoma is a neurodegenerative disorder affected by multiple factors, from which more than 60 million patients worldwide suffer. It is characterized by irreversible progressive loss of retinal ganglion cells (RGC) and distinct optic nerve head (ONH) deterioration, which is related to corresponding visual field loss.
- glaucoma
- matrix metalloproteinases
- trabecular meshwork
1. Introduction
| MMP Subtypes | MMP Number | Substrates |
|---|---|---|
| Collagenases [20,21,22][20][21][22] | MMP-1 | type I, II, III, VI, and X collagens; aggrecan; and entactin |
| MMP-8 | type I, II, and III collagens and aggrecan | |
| MMP-13 | type I, II, and III collagens | |
| Gelatinases [20,23,24][20][23][24] | MMP-2 | type I, IV, V, VII, X, and XI collagens; vitronectin; fibronectin; aggrecan; laminin; elastin; and tenascin C |
| MMP-9 | type IV, V, and XIV collagens; entactin; vitronectin; aggrecan; and elastin | |
| Stormelysins [20,25,26][20][25][26] | MMP-3 | type III, IV, IX, and X collagens; aggrecan; fibronectin; laminin; tenascin C; and vitronectin |
| MMP-10 | type IV collagen; aggrecan; and fibronectin | |
| MMP-11 | type IV collagen; fibronectin; laminin; and aggrecan | |
| Membrane-type MMPs [20,27][20][27] | MMP-14 | Activator of pro-MMP-2; type I, II, and III collagens; vitronectin; fibronectin; and laminin-1 |
| MMP-15 | Activator of pro-MMP-2; tenascin; fibronectin; and aggrecan | |
| MMP-16 | Activator of pro-MMP-2; fibronectin; and type III collagen |
|
| MMP-17 | Fibrin and cleaves pro-TNF-α | |
| MMP-24 | Activator of pro-MMP-2 | |
| MMP-25 | type IV collagen; gelatin; fibronectin; and fibrin | |
| Metalloelastases [20,26,28][20][26][28] | MMP-12 | type IV collagen; elastin; proteoglycan; fibronectin; laminin; and vitronectin |
| MMP-19 | cartilage oligomeric matrix protein and aggrecan | |
| MMP-20 | enamel matrix proteins | |
| MMP-28 | cartilage oligomeric matrix protein and aggrecan | |
| Other MMPs [20,26,29][20][26][29] | MMP-7 | aggrecan; fibronectin; laminin; and type IV collagen |
| MMP-26 | fibronectin; type IV collagen; fibrinogen; and gelatin |
2. Pathogenesis of Glaucoma concerning Matrix Metalloproteinases
2.1. Pathogenesis of Glaucoma Subtypes
Primary angle-closure glaucoma (PACG) is caused by compromised aqueous humor (AH) outflow and anterior chamber angle (ACA) obstruction. In most cases, the PACG angle narrows because of the relative pupillary block, which is related to displacement of the peripheral iris against the trabecular meshwork (TM) [45]. PACG prevalence differs according to ethnicity, sex, and family history. Studies have reported that PACG occurs three times more frequently in Asian populations than in European populations [46[46][47],47], and females are more prone to develop PACG [48,49][48][49]. Studies involving Chinese and Eskimos have shown that individuals with any first-degree relative with PACG are more susceptible to developing PACG [50,51][50][51]. Pseudoexfoliation glaucoma (XFG), which is caused by pseudoexfoliation syndrome (XFS), is the most commonly identifiable cause of open-angle glaucoma and is caused by pseudoexfoliation syndrome (XFS) [52]. XFS is an age-related syndrome characterized by the deposition of white scale-like substances in ocular tissues [53]. Scale-like substances are called exfoliation materials (XFM) and produce an abnormal accumulation of fibrillary elastic ECM [54]. The pathological events of XFG have not been fully confirmed; however, it is assumed that IOP is increased by AH outflow obstruction, which is induced by the deposition of XFM in the TM structure, finally leading to glaucomatous changes in the ONH [54]. Studies involving twins and first-degree relatives with XFS and loss of heterozygosity have indicated that XFG and XFS exhibit strong familial inheritance [55,56,57,58][55][56][57][58]. Juvenile open-angle glaucoma (JOAG) is an uncommon type of primary open-angle glaucoma (POAG), with a high prevalence in individuals aged 5–35 years. Individuals with JOAG follow autosomal dominant inheritance and have a strong family history of POAG [59]. Myocilin gene (MYOC) mutations account for approximately 10% of JOAG cases [31,60][31][60]. It has been suggested that MYOC mutations disrupt MMP and TIMP activities in the TM, causing pathological changes in the development of glaucoma [61,62][61][62].2.2. Matrix Metalloproteinases and Trabecular Meshwork with Glaucoma
Elevated IOP is one of the main risk factors for glaucoma [34,35][34][35]. IOP is defined as the difference between the production of aqueous humor (AH) in the ciliary body and AH drainage, mainly in trabecular meshwork (TM) and minorly in the uveoscleral pathway [63,64][63][64] (Figure 32). The TM generates the main outflow resistance for the AH, which is positioned at the iridocorneal angle, and its ECM is constantly remodeled by MMPs. ECM composition is continuously remodeled by selective ECM substrate degradation and production of new ECM substrates, including fibronectin, proteoglycans, collagens, and glycosaminoglycans, by TM cells. MMPs play a role in degrading specific ECM substrates [63,65][63][65]. Along with ECM modification, the geometry of the TM changes to regulate permeability via ciliary muscle contraction and form changes in the TM cells [66,67,68][66][67][68]. Many MMPs (MMP-1, -2, -3, -9, -12, and -14) and their local inhibitor TIMP-2 have been synthesized by TM cells [69,70][69][70]. Increasing MMP activity causes an elevated AH outflow rate, whereas inhibiting MMP activity decreases the AH outflow rate [71]. A recent study using a porcine model showed similar results, indicating that reduced MMP-2 and -9 activity is related to elevated IOP [72]. ECM composition changes can be observed when the AH outflow rate is pushed from equilibrium [73].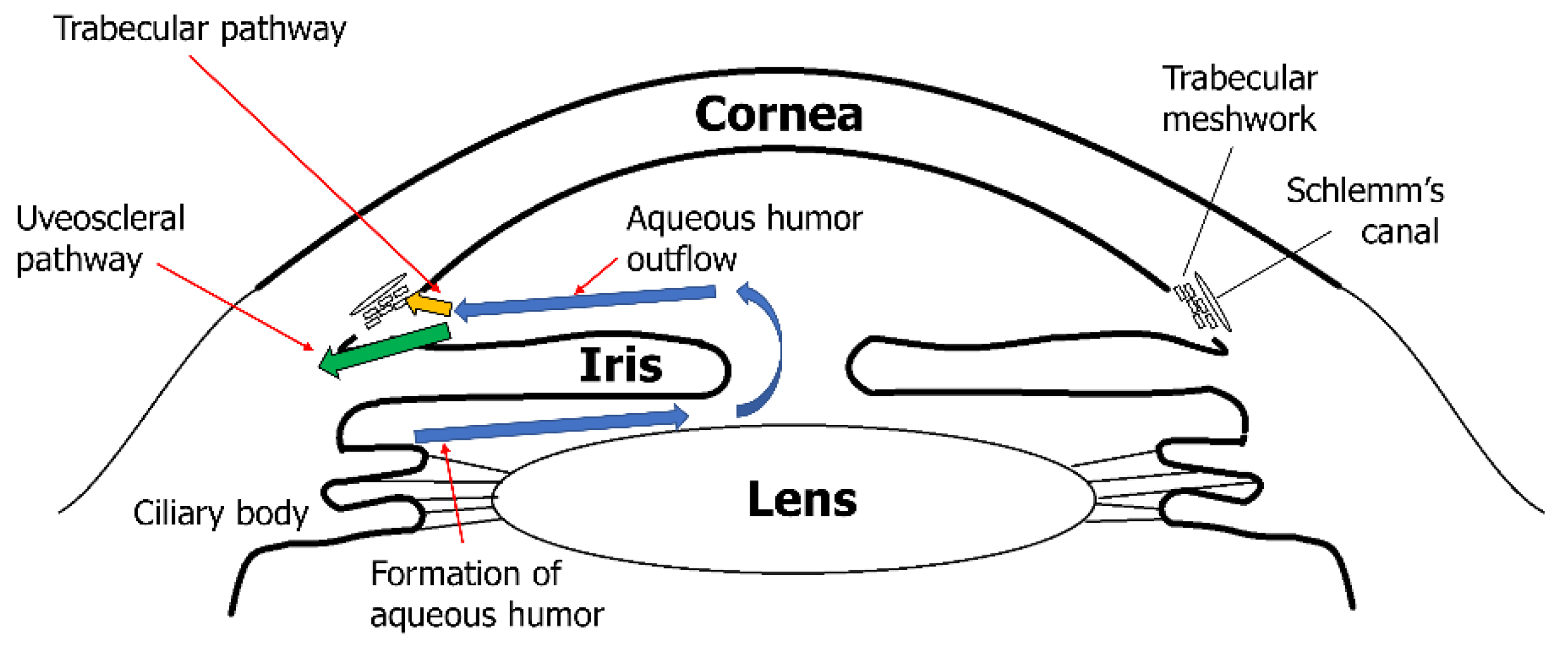
2.3. Matrix Metalloproteinases and Neuroretina with Glaucoma
MMPs play a major role in regulating AH outflow resistance by remodeling the ECM in the TM. However, MMPs are also factors that contribute to degenerative conditions in the posterior segment of the eye in glaucoma. Many MMPs and their inhibitors, TIMPs, are expressed in various neuronal and glial cells of the retina and the optic nerve. This expression may affect glaucomatous retinal and optic nerve degeneration by inducing apoptosis of retinal ganglion cells (RGC) and atrophic changes in the optic nerve [88,89,90,91,92,93][88][89][90][91][92][93]. However, the exact mechanism by which elevated IOP induces retinal degeneration and optic nerve atrophy remains unclear. Approximately 2 million RGC axons merge at a point in the posterior segment of the eye known as the optic nerve head (ONH). RGC axons at the ONH enter the neural canal and penetrate the Bruch’s membrane, choroid, and sclera [94]. As the RGC axon bundle passes through the ONH, a mesh-like structure called the lamina cribrosa (LC) supports the RGC axon bundles as they become the optic nerve [95,96][95][96]. In addition to the structural support of RGC axons, the LC has capillaries and glial cells within its structure that manage the ECM environment and provide nutritional support to RGC axons. As RGC axons forming the optic nerve are primarily supported by the LC, they are the main site within the rigid corneoscleral envelope, which is affected by the mechanical stress induced by increased IOP. Mechanical stress in the LC activates connective tissue remodeling cascades involving glial cells, LC cells, and scleral fibroblasts. Glial cells in the LC increase MMP secretion and modulate ECM structures to resemble glaucomatous environments. When ECM remodeling is complete, compression of RGC axons is relieved, and axoplasmic flow continues, which is affected by mechanical stress in the LC [97,98][97][98]. LC and glial cells are very sensitive to changes in mechanical stress that affect LC because they can sense mechanical stress by integrin receptors that are linked directly to the fibrillar ECM with cytoskeletons [99]. Many studies have investigated MMP and TIMP activities and expression in various glaucomatous animal models and humans with glaucomatous optic nerve changes. These studies suggest a mechanism by which MMPs affect glaucomatous optic nerve degeneration. In glaucomatous optic nerves, increased IOP induces mechanical stress in the LC, which is sensed by LC and glial cells. These cells secrete more cytokines (TGF-b1 and TNF-α) than normal cells do. Increased cytokine activity induces MMP-2 expression and ECM remodeling in the ONH. In addition, glial cells transform into reactive rather than quiescent forms and express MT1-MMP and MMP-1. MMP-1 causes continuous ECM degradation if it is not inhibited by TIMP-1, which is secreted by glial cells and RGC. If degradation continues, the LC cannot support RGC axon survival and may induce ONH excavation [41,91,92,97,98,100][41][91][92][97][98][100]. In patients with POAG and normal tension glaucoma (NTG), MMPs (MMP-1, -2, and -3) were enriched in the ONH [39,91,92][39][91][92] (Figure 43).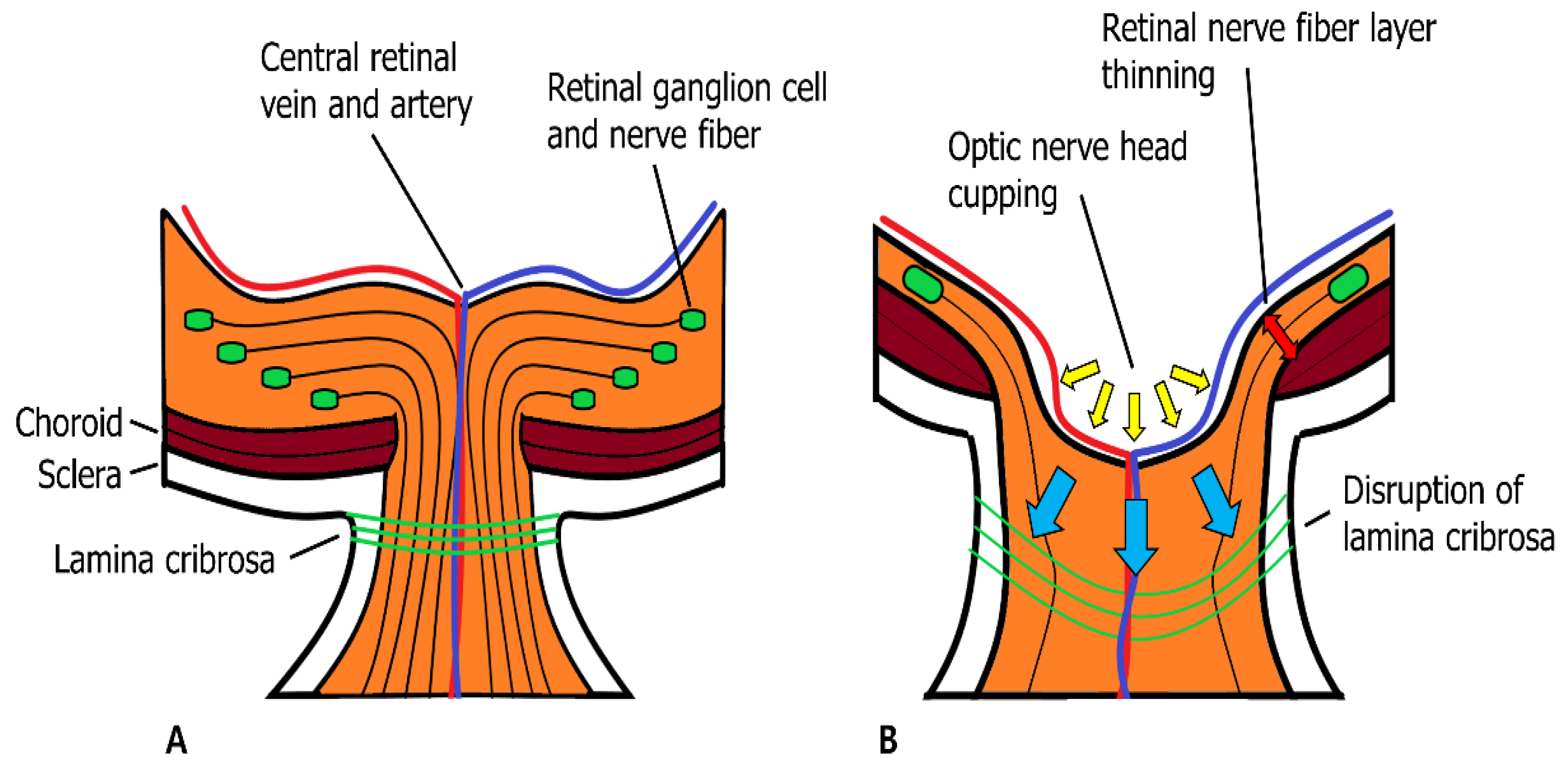
References
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516.
- Nagase, H.; Visse, R.; Murphy, G. Structure and Function of Matrix Metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573.
- McCawley, L.J.; Matrisian, L.M. Matrix Metalloproteinases: They’re Not Just for Matrix Anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540.
- Zhang, Y.; Klassen, H.J.; Tucker, B.A.; Perez, M.-T.R.; Young, M.J. CNS Progenitor Cells Promote a Permissive Environment for Neurite Outgrowth via a Matrix Metalloproteinase-2-Dependent Mechanism. J. Neurosci. 2007, 27, 4499–4506.
- Van Lint, P.; Libert, C. Chemokine and Cytokine Processing by Matrix Metalloproteinases and Its Effect on Leukocyte Migration and Inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381.
- McQuibban, G.A.; Gong, J.H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation Dampened by Gelatinase A Cleavage of Monocyte Chemoattractant Protein-3. Science 2000, 289, 1202–1206.
- Mannello, F.; Luchetti, F.; Falcieri, E.; Papa, S. Multiple Roles of Matrix Metalloproteinases during Apoptosis. Apoptosis 2005, 10, 19–24.
- Mott, J.D.; Werb, Z. Regulation of Matrix Biology by Matrix Metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564.
- Cauwe, B.; Van den Steen, P.E.; Opdenakker, G. The Biochemical, Biological, and Pathological Kaleidoscope of Cell Surface Substrates Processed by Matrix Metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 113–185.
- Tonti, G.A.; Mannello, F.; Cacci, E.; Biagioni, S. Neural Stem Cells at the Crossroads: MMPs May Tell the Way. Int. J. Dev. Biol. 2009, 53, 1–17.
- Chakraborti, S.; Mandal, M.; Das, S.; Mandal, A.; Chakraborti, T. Regulation of Matrix Metalloproteinases: An Overview. Mol. Cell Biochem. 2003, 253, 269–285.
- Spinale, F.G. Myocardial Matrix Remodeling and the Matrix Metalloproteinases: Influence on Cardiac Form and Function. Physiol. Rev. 2007, 87, 1285–1342.
- Rosenberg, G.A. Matrix Metalloproteinases and Their Multiple Roles in Neurodegenerative Diseases. Lancet Neurol. 2009, 8, 205–216.
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix Metalloproteinases Are Modifiers of Huntingtin Proteolysis and Toxicity in Huntington’s Disease. Neuron 2010, 67, 199–212.
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin Proteases and Their Inhibitors: Foes or Friends in Nervous System Physiology? J. Neurosci. 2010, 30, 15337–15357.
- Yong, V.W. Metalloproteinases: Mediators of Pathology and Regeneration in the CNS. Nat. Rev. Neurosci. 2005, 6, 931–944.
- Fujioka, H.; Dairyo, Y.; Yasunaga, K.; Emoto, K. Neural Functions of Matrix Metalloproteinases: Plasticity, Neurogenesis, and Disease. Biochem. Res. Int. 2012, 2012, 789083.
- Klein, T.; Bischoff, R. Physiology and Pathophysiology of Matrix Metalloproteases. Amino Acids 2011, 41, 271–290.
- Nagase, H.; Woessner, J.F. Matrix Metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494.
- de Almeida, L.G.N.; Thode, H.; Eslambolchi, Y.; Chopra, S.; Young, D.; Gill, S.; Devel, L.; Dufour, A. Matrix Metalloproteinases: From Molecular Mechanisms to Physiology, Pathophysiology, and Pharmacology. Pharmacol. Rev. 2022, 74, 712–768.
- Ratnikov, B.I.; Cieplak, P.; Gramatikoff, K.; Pierce, J.; Eroshkin, A.; Igarashi, Y.; Kazanov, M.; Sun, Q.; Godzik, A.; Osterman, A.; et al. Basis for Substrate Recognition and Distinction by Matrix Metalloproteinases. Proc. Natl. Acad. Sci. USA 2014, 111, E4148–E4155.
- Alipour, H.; Raz, A.; Zakeri, S.; Dinparast Djadid, N. Therapeutic Applications of Collagenase (Metalloproteases): A Review. Asian Pac. J. Trop. Biomed. 2016, 6, 975–981.
- Hannocks, M.-J.; Zhang, X.; Gerwien, H.; Chashchina, A.; Burmeister, M.; Korpos, E.; Song, J.; Sorokin, L. The Gelatinases, MMP-2 and MMP-9, as Fine Tuners of Neuroinflammatory Processes. Matrix Biol. 2019, 75–76, 102–113.
- Nikolov, A.; Popovski, N. Role of Gelatinases MMP-2 and MMP-9 in Healthy and Complicated Pregnancy and Their Future Potential as Preeclampsia Biomarkers. Diagnostics 2021, 11, 480.
- Raeeszadeh-Sarmazdeh, M.; Coban, M.; Mahajan, S.; Hockla, A.; Sankaran, B.; Downey, G.P.; Radisky, D.C.; Radisky, E.S. Engineering of Tissue Inhibitor of Metalloproteinases TIMP-1 for Fine Discrimination between Closely Related Stromelysins MMP-3 and MMP-10. J. Biol. Chem. 2022, 298, 101654.
- Kandhwal, M.; Behl, T.; Singh, S.; Sharma, N.; Arora, S.; Bhatia, S.; Al-Harrasi, A.; Sachdeva, M.; Bungau, S. Role of Matrix Metalloproteinase in Wound Healing. Am. J. Transl. Res. 2022, 14, 4391–4405.
- Fogarasi, M.; Dima, S. The Catalytic Domain Mediates Homomultimerization of MT1-MMP and the Prodomain Interferes with MT1-MMP Oligomeric Complex Assembly. Biomolecules 2022, 12, 1145.
- Chuliá-Peris, L.; Carreres-Rey, C.; Gabasa, M.; Alcaraz, J.; Carretero, J.; Pereda, J. Matrix Metalloproteinases and Their Inhibitors in Pulmonary Fibrosis: EMMPRIN/CD147 Comes into Play. Int. J. Mol. Sci. 2022, 23, 6894.
- Loreto, C.; Polizzi, A.; Filetti, V.; Pannone, G.; Dos Santos, J.N.; Venezia, P.; Leonardi, R.; Isola, G. Expression of Matrix Metalloproteinases 7 and 9, Desmin, Alpha-Smooth Muscle Actin and Caldesmon, in Odontogenic Keratocyst Associated with NBCCS, Recurrent and Sporadic Keratocysts. Biomolecules 2022, 12, 775.
- Weinreb, R.N.; Robinson, M.R.; Dibas, M.; Stamer, W.D. Matrix Metalloproteinases and Glaucoma Treatment. J. Ocul. Pharmacol. Ther. 2020, 36, 208–228.
- Kwon, Y.H.; Fingert, J.H.; Kuehn, M.H.; Alward, W.L.M. Primary Open-Angle Glaucoma. N. Engl. J. Med. 2009, 360, 1113–1124.
- Weinreb, R.N.; Khaw, P.T. Primary Open-Angle Glaucoma. Lancet 2004, 363, 1711–1720.
- Quigley, H.A. Number of People with Glaucoma Worldwide. Br. J. Ophthalmol. 1996, 80, 389–393.
- Heijl, A. Reduction of Intraocular Pressure and Glaucoma Progression: Results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002, 120, 1268.
- Iwase, A.; Suzuki, Y.; Araie, M.; Yamamoto, T.; Abe, H.; Shirato, S.; Kuwayama, Y.; Mishima, H.; Shimizu, H.; Tomita, G. The Prevalence of Primary Open-Angle Glaucoma in Japanese: The Tajimi Study. Ophthalmology 2004, 111, 1641–1648.
- Bouhenni, R.A.; Dunmire, J.; Sewell, A.; Edward, D.P. Animal Models of Glaucoma. J. Biomed. Biotechnol. 2012, 2012, 692609.
- Johnson, T.V.; Tomarev, S.I. Rodent Models of Glaucoma. Brain Res. Bull. 2010, 81, 349–358.
- Guo, Y.; Cepurna, W.O.; Dyck, J.A.; Doser, T.A.; Johnson, E.C.; Morrison, J.C. Retinal Cell Responses to Elevated Intraocular Pressure: A Gene Array Comparison between the Whole Retina and Retinal Ganglion Cell Layer. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3003.
- Myers, J.S. Retinal Ganglion Cell Apoptosis in Glaucoma Is Related to Intraocular Pressure and IOP-Induced Effects on Extracellular Matrix. Yearb. Ophthalmol. 2006, 2006, 83–85.
- De Groef, L.; Van Hove, I.; Dekeyster, E.; Stalmans, I.; Moons, L. MMPs in the Trabecular Meshwork: Promising Targets for Future Glaucoma Therapies? Investig. Ophthalmol. Vis. Sci. 2013, 54, 7756–7763.
- De Groef, L.; Van Hove, I.; Dekeyster, E.; Stalmans, I.; Moons, L. MMPs in the Neuroretina and Optic Nerve: Modulators of Glaucoma Pathogenesis and Repair? Investig. Ophthalmol. Vis. Sci. 2014, 55, 1953.
- Shima, I.; Katsuda, S.; Ueda, Y.; Takahashi, N.; Sasaki, H. Expression of Matrix Metalloproteinases in Wound Healing after Glaucoma Filtration Surgery in Rabbits. Ophthalmic Res. 2007, 39, 315–324.
- Zaleska-Żmijewska, A.; Strzemecka, E.; Wawrzyniak, Z.M.; Szaflik, J.P. Extracellular MMP-9-Based Assessment of Ocular Surface Inflammation in Patients with Primary Open-Angle Glaucoma. J. Ophthalmol. 2019, 2019, 1240537.
- Hussain, A.A.; Lee, Y.; Zhang, J.-J.; Marshall, J. Characterization of the Gelatinase System of the Laminar Human Optic Nerve, and Surrounding Annulus of Bruch’s Membrane, Choroid, and Sclera. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2358.
- Foster, P.J.; Buhrmann, R.; Quigley, H.A.; Johnson, G.J. The Definition and Classification of Glaucoma in Prevalence Surveys. Br. J. Ophthalmol. 2002, 86, 238–242.
- Dandona, L.; Dandona, R.; Mandal, P.; Srinivas, M.; John, R.K.; McCarty, C.A.; Rao, G.N. Angle-Closure Glaucoma in an Urban Population in Southern India. The Andhra Pradesh Eye Disease Study. Ophthalmology 2000, 107, 1710–1716.
- Foster, P.J.; Oen, F.T.; Machin, D.; Ng, T.P.; Devereux, J.G.; Johnson, G.J.; Khaw, P.T.; Seah, S.K. The Prevalence of Glaucoma in Chinese Residents of Singapore: A Cross-Sectional Population Survey of the Tanjong Pagar District. Arch. Ophthalmol. 2000, 118, 1105–1111.
- Vajaranant, T.S.; Nayak, S.; Wilensky, J.T.; Joslin, C.E. Gender and Glaucoma: What We Know and What We Need to Know. Curr. Opin. Ophthalmol. 2010, 21, 91–99.
- Foster, P.J.; Alsbirk, P.H.; Baasanhu, J.; Munkhbayar, D.; Uranchimeg, D.; Johnson, G.J. Anterior Chamber Depth in Mongolians: Variation with Age, Sex, and Method of Measurement. Am. J. Ophthalmol. 1997, 124, 53–60.
- Alsbirk, P.H. Anterior Chamber Depth and Primary Angle-Closure Glaucoma. I. An Epidemiologic Study in Greenland Eskimos. Acta Ophthalmol. 1975, 53, 89–104.
- He, M.; Wang, D.; Zheng, Y.; Zhang, J.; Yin, Q.; Huang, W.; Mackey, D.A.; Foster, P.J. Heritability of Anterior Chamber Depth as an Intermediate Phenotype of Angle-Closure in Chinese: The Guangzhou Twin Eye Study. Investig. Ophthalmol. Vis. Sci. 2008, 49, 81–86.
- Ritch, R. Exfoliation Syndrome-the Most Common Identifiable Cause of Open-Angle Glaucoma. J. Glaucoma 1994, 3, 176–177.
- Zenkel, M.; Schlötzer-Schrehardt, U. The Composition of Exfoliation Material and the Cells Involved in Its Production. J. Glaucoma 2014, 23, S12–S14.
- Schlötzer-Schrehardt, U.; Naumann, G.O. Ocular and Systemic Pseudoexfoliation Syndrome. Am. J. Ophthalmol. 2006, 141, 921–937.
- Gottfredsdottir, M.S.; Sverrisson, T.; Musch, D.C.; Stefansson, E. Chronic Open-Angle Glaucoma and Associated Ophthalmic Findings in Monozygotic Twins and Their Spouses in Iceland. J. Glaucoma 1999, 8, 134–139.
- Aasved, H. Study of Relatives of Persons with Fibrillopathia Epitheliocapsularis (Pseudoexfoliation of the Lens Capsule). Acta Ophthalmol. 1975, 53, 879–886.
- Kozobolis, V.P.; Detorakis, E.T.; Sourvinos, G.; Pallikaris, I.G.; Spandidos, D.A. Loss of Heterozygosity in Pseudoexfoliation Syndrome. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1255–1260.
- Challa, P. Genetics of Pseudoexfoliation Syndrome. Curr. Opin. Ophthalmol. 2009, 20, 88–91.
- Turalba, A.V.; Chen, T.C. Clinical and Genetic Characteristics of Primary Juvenile-Onset Open-Angle Glaucoma (JOAG). Semin. Ophthalmol. 2008, 23, 19–25.
- Fingert, J.H.; Stone, E.M.; Sheffield, V.C.; Alward, W.L.M. Myocilin Glaucoma. Surv. Ophthalmol. 2002, 47, 547–561.
- Ashworth Briggs, E.L.; Toh, T.; Eri, R.; Hewitt, A.W.; Cook, A.L. TIMP1, TIMP2, and TIMP4 Are Increased in Aqueous Humor from Primary Open Angle Glaucoma Patients. Mol. Vis. 2015, 21, 1162–1172.
- Peters, J.C.; Bhattacharya, S.; Clark, A.F.; Zode, G.S. Increased Endoplasmic Reticulum Stress in Human Glaucomatous Trabecular Meshwork Cells and Tissues. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3860–3868.
- Vranka, J.A.; Kelley, M.J.; Acott, T.S.; Keller, K.E. Extracellular Matrix in the Trabecular Meshwork: Intraocular Pressure Regulation and Dysregulation in Glaucoma. Exp. Eye Res. 2015, 133, 112–125.
- Tian, B.; Gabelt, B.T.; Geiger, B.; Kaufman, P.L. The Role of the Actomyosin System in Regulating Trabecular Fluid Outflow. Exp. Eye Res. 2009, 88, 713–717.
- Tane, N.; Dhar, S.; Roy, S.; Pinheiro, A.; Ohira, A.; Roy, S. Effect of Excess Synthesis of Extracellular Matrix Components by Trabecular Meshwork Cells: Possible Consequence on Aqueous Outflow. Exp. Eye Res. 2007, 84, 832–842.
- Oh, D.-J.; Martin, J.L.; Williams, A.J.; Russell, P.; Birk, D.E.; Rhee, D.J. Effect of Latanoprost on the Expression of Matrix Metalloproteinases and Their Tissue Inhibitors in Human Trabecular Meshwork Cells. Invest. Ophthalmol. Vis. Sci. 2006, 47, 3887–3895.
- Keller, K.E.; Acott, T.S. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J. Ocul. Biol. 2013, 1, 3.
- Tamm, E.R. The Trabecular Meshwork Outflow Pathways: Structural and Functional Aspects. Exp. Eye Res. 2009, 88, 648–655.
- Alexander, J.P.; Samples, J.R.; Van Buskirk, E.M.; Acott, T.S. Expression of Matrix Metalloproteinases and Inhibitor by Human Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 1991, 32, 172–180.
- Kelley, M.J.; Rose, A.Y.; Song, K.; Chen, Y.; Bradley, J.M.; Rookhuizen, D.; Acott, T.S. Synergism of TNF and IL-1 in the Induction of Matrix Metalloproteinase-3 in Trabecular Meshwork. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2634–2643.
- Bradley, J.; Vranka, J.; Colvis, C.; Conger, D.; Alexander, J.; Fisk, A.; Samples, J.; Acott, T. Effect of Matrix Metalloproteinases Activity on Outflow in Perfused Human Organ Culture. Investig. Ophthalmol. Vis. Sci. 1999, 39, 2649–2658.
- Snider, E.J.; Hardie, B.A.; Li, Y.; Gao, K.; Splaine, F.; Kim, R.K.; Vannatta, R.T.; Read, A.T.; Ethier, C.R. A Porcine Organ-Culture Glaucoma Model Mimicking Trabecular Meshwork Damage Using Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2021, 62, 18.
- Acott, T.S.; Kelley, M.J. Extracellular Matrix in the Trabecular Meshwork. Exp. Eye Res. 2008, 86, 543–561.
- Bradley, J.M.B.; Kelley, M.J.; Rose, A.; Acott, T.S. Signaling Pathways Used in Trabecular Matrix Metalloproteinase Response to Mechanical Stretch. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5174–5181.
- Bradley, J.M.B.; Kelley, M.J.; Zhu, X.; Anderssohn, A.M.; Alexander, J.P.; Acott, T.S. Effects of Mechanical Stretching on Trabecular Matrix Metalloproteinases. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1505–1513.
- Luna, C.; Li, G.; Liton, P.B.; Epstein, D.L.; Gonzalez, P. Alterations in Gene Expression Induced by Cyclic Mechanical Stress in Trabecular Meshwork Cells. Mol. Vis. 2009, 15, 534–544.
- WuDunn, D. The Effect of Mechanical Strain on Matrix Metalloproteinase Production by Bovine Trabecular Meshwork Cells. Curr. Eye Res. 2001, 22, 394–397.
- Fleenor, D.L.; Pang, I.-H.; Clark, A.F. Involvement of AP-1 in Interleukin-1α–Stimulated MMP-3 Expression in Human Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3494–3501.
- Aga, M.; Bradley, J.M.; Keller, K.E.; Kelley, M.J.; Acott, T.S. Specialized Podosome- or Invadopodia-like Structures (PILS) for Focal Trabecular Meshwork Extracellular Matrix Turnover. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5353–5365.
- Guo, M.-S.; Wu, Y.-Y.; Liang, Z.-B. Hyaluronic Acid Increases MMP-2 and MMP-9 Expressions in Cultured Trabecular Meshwork Cells from Patients with Primary Open-Angle Glaucoma. Mol. Vis. 2012, 18, 1175–1181.
- Knepper, P.A.; Goossens, W.; Hvizd, M.; Palmberg, P.F. Glycosaminoglycans of the Human Trabecular Meshwork in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1360–1367.
- Schlötzer-Schrehardt, U.; Lommatzsch, J.; Küchle, M.; Konstas, A.G.P.; Naumann, G.O.H. Matrix Metalloproteinases and Their Inhibitors in Aqueous Humor of Patients with Pseudoexfoliation Syndrome/Glaucoma and Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1117–1125.
- Määttä, M.; Tervahartiala, T.; Harju, M.; Airaksinen, J.; Autio-Harmainen, H.; Sorsa, T. Matrix Metalloproteinases and Their Tissue Inhibitors in Aqueous Humor of Patients with Primary Open-Angle Glaucoma, Exfoliation Syndrome, and Exfoliation Glaucoma. J. Glaucoma 2005, 14, 64–69.
- Gaballa, S.A.; Kompella, U.B.; Elgarhy, O.; Alqahtani, A.M.; Pierscionek, B.; Alany, R.G.; Abdelkader, H. Corticosteroids in Ophthalmology: Drug Delivery Innovations, Pharmacology, Clinical Applications, and Future Perspectives. Drug Deliv. Transl. Res. 2021, 11, 866–893.
- Phulke, S.; Kaushik, S.; Kaur, S.; Pandav, S. Steroid-Induced Glaucoma: An Avoidable Irreversible Blindness. J. Curr. Glaucoma Pract. 2017, 11, 67–72.
- Mohd Nasir, N.A.; Agarwal, R.; Krasilnikova, A.; Sheikh Abdul Kadir, S.H.; Iezhitsa, I. Effect of Dexamethasone on the Expression of MMPs, Adenosine A1 Receptors and NFKB by Human Trabecular Meshwork Cells. J. Basic Clin. Physiol. Pharmacol. 2020, 31, 20190373.
- Saadat, F.; Raji, A.; Eslami, K.Z.M.B.; Pezeshki, M.; Aalizadeh, M.R.K.N. Alteration in Matrix Metalloproteinases (MMPS) Activity in Fibroblast Cell Line by Dexamethasone: A Possible Mechanism in Corticosteroid-Induced Glaucoma. Iran. J. Allergy Asthma Immunol. 2003, 2, 145–148.
- Hofmaier, F.; Hauck, S.M.; Amann, B.; Degroote, R.L.; Deeg, C.A. Changes in Matrix Metalloproteinase Network in a Spontaneous Autoimmune Uveitis Model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2314–2320.
- Agapova, O.A.; Kaufman, P.L.; Lucarelli, M.J.; Gabelt, B.T.; Hernandez, M.R. Differential Expression of Matrix Metalloproteinases in Monkey Eyes with Experimental Glaucoma or Optic Nerve Transection. Brain Res. 2003, 967, 132–143.
- Sun, M.-H.; Chen, K.-J.; Tsao, Y.-P.; Kao, L.-Y.; Han, W.-H.; Lin, K.-K.; Pang, J.-H.S. Down-Regulation of Matrix Metalloproteinase-9 by Pyrrolidine Dithiocarbamate Prevented Retinal Ganglion Cell Death after Transection of Optic Nerve in Rats. Curr. Eye Res. 2011, 36, 1053–1063.
- Agapova, O.A.; Ricard, C.S.; Salvador-Silva, M.; Hernandez, M.R. Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Optic Nerve Head Astrocytes. Glia 2001, 33, 205–216.
- Yan, X.; Tezel, G.; Wax, M.B.; Edward, D.P. Matrix Metalloproteinases and Tumor Necrosis Factor Alpha in Glaucomatous Optic Nerve Head. Arch. Ophthalmol. 2000, 118, 666–673.
- Webster, L.; Chignell, A.H.; Limb, G.A. Predominance of MMP-1 and MMP-2 in Epiretinal and Subretinal Membranes of Proliferative Vitreoretinopathy. Exp. Eye Res. 1999, 68, 91–98.
- Burgoyne, C.F.; Downs, J.C. Premise and Prediction—How Optic Nerve Head Biomechanics Underlies the Susceptibility and Clinical Behavior of the Aged Optic Nerve Head. J. Glaucoma 2008, 17, 318–328.
- Emery, J.M.; Landis, D.; Paton, D.; Boniuk, M.; Craig, J.M. The Lamina Cribrosa in Normal and Glaucomatous Human Eyes. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1974, 78, 290–297.
- Downs, J.C.; Girkin, C.A. Lamina Cribrosa in Glaucoma. Curr. Opin. Ophthalmol. 2017, 28, 113–119.
- Quill, B.; Docherty, N.G.; Clark, A.F.; O’Brien, C.J. The Effect of Graded Cyclic Stretching on Extracellular Matrix–Related Gene Expression Profiles in Cultured Primary Human Lamina Cribrosa Cells. Invesigt. Ophthalmol. Vis. Sci. 2011, 52, 1908.
- Kirwan, R.P.; Crean, J.K.; Fenerty, C.H.; Clark, A.F.; O’Brien, C.J. Effect of Cyclical Mechanical Stretch and Exogenous Transforming Growth Factor-beta1 on Matrix Metalloproteinase-2 Activity in Lamina Cribrosa Cells from the Human Optic Nerve Head. J. Glaucoma 2004, 13, 327–334.
- Morrison, J.C. Integrins in the Optic Nerve Head: Potential Roles in Glaucomatous Optic Neuropathy (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2006, 104, 453–477.
- Akhter, N.; Nix, M.; Abdul, Y.; Singh, S.; Husain, S. Delta-Opioid Receptors Attenuate TNF-α–Induced MMP-2 Secretion from Human ONH Astrocytes. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6605–6611.
- Albon, J.; Karwatowski, W.S.; Avery, N.; Easty, D.L.; Duance, V.C. Changes in the Collagenous Matrix of the Aging Human Lamina Cribrosa. Br. J. Ophthalmol. 1995, 79, 368–375.
- Morrison, J.C.; Jerdan, J.A.; Dorman, M.E.; Quigley, H.A. Structural Proteins of the Neonatal and Adult Lamina Cribrosa. Arch. Ophthalmol. 1989, 107, 1220–1224.
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic Nerve Damage in Human Glaucoma. II. The Site of Injury and Susceptibility to Damage. Arch. Ophthalmol. 1981, 99, 635–649.
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Hart, R.T. Three-Dimensional Reconstruction of Normal and Early Glaucoma Monkey Optic Nerve Head Connective Tissues. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4388–4399.
- Yang, H.; Downs, J.C.; Girkin, C.; Sakata, L.; Bellezza, A.; Thompson, H.; Burgoyne, C.F. 3-D Histomorphometry of the Normal and Early Glaucomatous Monkey Optic Nerve Head: Lamina Cribrosa and Peripapillary Scleral Position and Thickness. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4597–4607.
- Halfter, W.; Willem, M.; Mayer, U. Basement Membrane-Dependent Survival of Retinal Ganglion Cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1000–1009.
- Zhang, X.; Cheng, M.; Chintala, S.K. Optic Nerve Ligation Leads to Astrocyte-Associated Matrix Metalloproteinase-9 Induction in the Mouse Retina. Neurosci. Lett. 2004, 356, 140–144.
- Zhang, X.; Cheng, M.; Chintala, S.K. Kainic Acid-Mediated Upregulation of Matrix Metalloproteinase-9 Promotes Retinal Degeneration. Invest. Ophthalmol. Vis. Sci. 2004, 45, 2374–2383.
- Zhang, X.; Chintala, S.K. Influence of Interleukin-1 Beta Induction and Mitogen-Activated Protein Kinase Phosphorylation on Optic Nerve Ligation-Induced Matrix Metalloproteinase-9 Activation in the Retina. Exp. Eye Res. 2004, 78, 849–860.
- Manabe, S.-I.; Gu, Z.; Lipton, S.A. Activation of Matrix Metalloproteinase-9 via Neuronal Nitric Oxide Synthase Contributes to NMDA-Induced Retinal Ganglion Cell Death. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4747–4753.
- Chintala, S.K.; Zhang, X.; Austin, J.S.; Fini, M.E. Deficiency in Matrix Metalloproteinase Gelatinase B (MMP-9) Protects against Retinal Ganglion Cell Death after Optic Nerve Ligation. J. Biol. Chem. 2002, 277, 47461–47468.
- Santos, A.R.C.; Corredor, R.G.; Obeso, B.A.; Trakhtenberg, E.F.; Wang, Y.; Ponmattam, J.; Dvoriantchikova, G.; Ivanov, D.; Shestopalov, V.I.; Goldberg, J.L.; et al. Β1 Integrin-Focal Adhesion Kinase (FAK) Signaling Modulates Retinal Ganglion Cell (RGC) Survival. PLoS ONE 2012, 7, e48332.
- Zhang, X.; Sakamoto, T.; Hata, Y.; Kubota, T.; Hisatomi, T.; Murata, T.; Ishibashi, T.; Inomata, H. Expression of Matrix Metalloproteinases and Their Inhibitors in Experimental Retinal Ischemia-Reperfusion Injury in Rats. Exp. Eye Res. 2002, 74, 577–584.
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-Nitrosylation of Matrix Metalloproteinases: Signaling Pathway to Neuronal Cell Death. Science 2002, 297, 1186–1190.
- Kido, N.; Inatani, M.; Honjo, M.; Yoneda, S.; Hara, H.; Miyawaki, N.; Honda, Y.; Tanihara, H. Dual Effects of Interleukin-1beta on N-Methyl-D-Aspartate-Induced Retinal Neuronal Death in Rat Eyes. Brain Res. 2001, 910, 153–162.