Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Hester van Mourik.
Cathepsins are lysosomal proteases that are essential to maintain cellular physiological homeostasis and are involved in multiple processes, such as immune and energy regulation. Cathepsins are also involved in pathological situations, especially when they are secreted and enter the extracellular space. Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and it is the third leading cause of cancer-related deaths worldwide. Cathepsins are involved in the pathology of HCC through acting in proliferation, apoptosis, angiogenesis, invasion and metastasis, metabolism and immunity.
- NASH
- HCC
- cathepsins
- cancer hallmarks
1. Introduction
The term “cathepsin” (derived from the Greek word “Καθεψίνη”, which means to “boil down” or “digest”) was first coined in 1929 by Nobel Prize winner Richard Willstätter to describe all proteases that degrade proteins [1]. Cathepsins are now recognized as proteases that are predominantly active in the endo-lysosomal compartment [2]. These proteases are subdivided into three distinguished families based on which amino acid is present at their active site, i.e., the serine proteases (cathepsin A, G), the aspartic proteases (cathepsin D, E), and the cysteine proteases (cathepsin B, C, F, H, K, L, O, S, V, X, W) [3]. The most abundant cathepsins are cathepsin B, D, and L (CTSB, CTSD, CTSL respectively) [4]. Although most cathepsins are ubiquitously expressed throughout the body, some show tissue-specific expression [5]. This tissue-specific expression is important since the function of cathepsin can differ per tissue type.
2. Mechanisms of Cathepsins in HCC
Cathepsins are ubiquitously expressed within various tissue types and especially in the context of tumor biology, often secreted into the tumor microenvironment (TME). Many cell types, including macrophages and fibroblasts, contribute to the pool of cathepsins in the TME [3]. Here, wresearchers describe the mechanisms by which cathepsins operate in HCC by using the hallmarks of cancer concept [16,17,18][6][7][8]. Hereby, the hallmarks where the involvement of cathepsins is only marginally or not described (evading growth suppressors, enabling replicative immortality, unlocking phenotype plasticity) are left out.
2.1. Cathepsins Sustain Proliferative Signaling through the PI3K/Akt/mTOR Signaling Pathway in HCC
Cancer cells have developed the ability to uncontrollably proliferate without the need for external proliferative signals. This ability is modulated by pathways that are involved in cell proliferation and cell growth, thereby moving away from the carefully orchestrated homeostasis of normal cells [17][7].
A plethora of studies have pointed towards a role for cathepsins in the progression of HCC. In vitro overexpression of CTSB and CTSL in various HCC cell lines stimulated cell growth and proliferation in at least two independent studies [13,72][9][10]. In line, the silencing of CTSB, CTSL, and CTSS in several HCC cells resulted in the reverse, i.e., decreased cell proliferation [13,72,73][9][10][11]. Similarly, it was demonstrated in vivo that the injection of CTSL-overexpressing HCC (MHCC-97-H) cells into mice resulted in increased growth and tumor weight compared to the control [13][9]. Furthermore, when mice were injected with HCC (HepG2) cells that were silenced in CTSB, tumor size and weight decreased [72][10].
The involvement of cathepsins in cancer proliferation is related to its ability to affect intracellular signaling pathways. Several studies demonstrated that cathepsins upregulate the PI3K/Akt/mTOR pathway (crucial in cell growth, proliferation, and survival [74][12]) through the phosphorylation of the kinase Akt (pAkt) [72,75,76][10][13][14]. In HCC (BEL-7402) cells overexpressing CTSB, the levels of pAkt were elevated compared to the control. Likewise, HCC (HepG2) cells with downregulated CTSB displayed reduced levels of pAkt [72][10]. The decrease in pAkt was also observed in acute myeloid leukemia cells, upon silencing of CTSB [75][13]. Other studies demonstrated that mice with breast cancer that had a CTSD knockout (MMTV-PyMT-cre; CTSD−/−) elicited impaired mTOR signaling, which is an important downstream target of pAkt [76][14], suggesting that cathepsins are involved in this crucial cell survival and growth pathway. Although no direct evidence is available regarding the involvement of CTSS and CTSL in the PI3K/Akt/mTOR pathway in HCC proliferation, the observation that in glioblastoma cells, the inhibition of CTSS reduces the phosphorylation of Akt [77][15] suggests that the involvement of cathepsins in this pathway is common in different types of cancers, including HCC.
Overall, available data suggest intracellular cathepsins are active players in uncontrolled cancer cell proliferation, specifically by interfering with the pro-survival and proliferation PI3K/Akt/mTOR pathway.
2.2. Elevated Levels of CTSB and CTSD Result in Apoptosis Whereas Elevated CTSS Levels Lead to Resistance of Apoptosis in HCC
Tumor cells have acquired the ability to resist programmed cell death or apoptosis and to uncontrollably expand in number. The role of cathepsins in these processes has been described in tumors of different origin, including HCC. While intracellular CTSB and CTSD have been demonstrated to be pro-apoptotic, intracellular CTSS has been found to be anti-apoptotic in HCC (thus, tumor promoting).
The importance of lysosomal disruption and the subsequent release of CTSB for apoptosis in HCC (Huh-7, Hep3B) cells has been elegantly demonstrated by Guicciardi et al. [78][16]. Their data show that apoptosis is triggered by the TNF-related apoptosis-inducing ligand (TRAIL) pathway (Figure 1). Activation of this pathway eventually leads to the destabilization of the lysosome and to the lysosomal release of CTSB [78][16]. In addition, TNF-α combined with cycloheximide increased the concentration of sphingosine inside the lysosome, which in turn triggered LMP in rat (HTC), murine (1c1c7), and human HCC (SK-HEP-1) cells, resulting in the release of CTSB and CTSD [79,80][17][18]. In cancers, including in HCC, this pathway is often prevented by the upregulation of the anti-apoptotic protein cFLIP (cellular FLICE/caspase-8 inhibitory protein), since this stabilizes the lysosome [78][16].
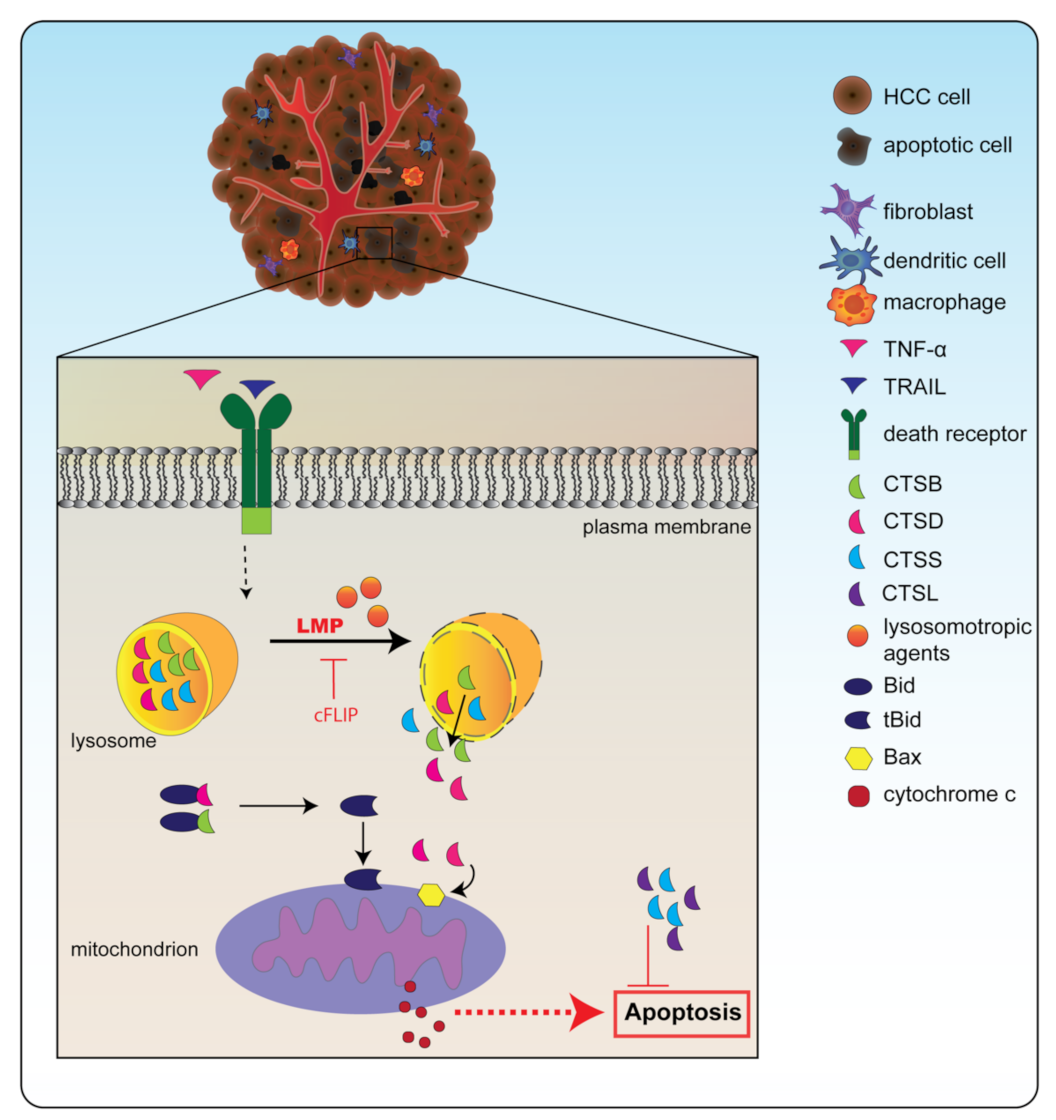
Figure 1. Cathepsins are involved in apoptosis. Destabilization of the lysosome through factors such as TNF-related apoptosis-inducing ligand (TRAIL) or tumor-necrosis factor α (TNF-α) combined with a lysosomotropic agent leads to lysosomal membrane permeabilization (LMP). Through LMP, cathepsins can be released from the lysosome into the cytosol. In the cytosol, cathepsin B (CTSB) and cathepsin D (CTSD) can cleave pro-apoptotic factor Bid into its active version truncated (t)Bid. In addition, these cathepsins can stimulate Bax insertion into the mitochondrial membrane. Together, this leads to the release of cytochrome c, which can induce the caspase cascade, eventually culminating in apoptosis. Cathepsin S (CTSS) and cathepsin L (CTSL), however, can inhibit apoptosis and therefore when they are upregulated, cancer cells can resist this form of cell death.
Strikingly, permeabilization of the lysosomal membrane in HCC cells resulted in the release of CTSB and CTSD into the cytosol of HCC cells, where they then cleaved essential apoptotic substrates [78[16][17][19],79,81], including the pro-apoptotic Bid, which then turns into its truncated version, tBid [42,79,82,83][17][20][21][22]. In addition, it was determined that upon lysosomal CTSD release in HCC (BEL-7404) cells, the pro-apoptotic protein Bax was inserted into the mitochondrial membrane [83,84][22][23]. tBid formation and mitochondrial Bax insertion result in the release of mitochondrial cytochrome c and subsequently caspase activation, which leads to apoptosis [82][21].
While the abovementioned studies described a role for CTSB in HCC, CTSB also seems to play a role in non-cancerous liver cells. In non-cancerous rat hepatocytes, CTSB inhibition reduced apoptosis activity [85][24]. In addition, cells in CTSB−/− mice were resilient to TNF-α-induced apoptosis, suggesting the importance of this protease in apoptosis [86][25].
In contrast to CTSD and CTSB stimulating apoptosis, CTSS actually inhibits apoptosis and thereby promotes tumor progression. CTSS is usually highly expressed in immune cells but also in malignant cells, including HCC cells [4]. CTSS knockdown in HCC (MHCC-97-H) cells resulted in higher concentrations of cleaved caspase 3 and increased apoptosis [73,87][11][26]. Similar effects on the inhibition of CTSS-induced apoptosis were observed in human glioblastoma and renal carcinoma cells [77,88,89][15][27][28]. Hence, these results suggest that CTSS appears to be crucial in preventing apoptosis and that targeting it may be an interesting approach to tackle this evasion of apoptosis in cancer cells.
While there is no information available on the role of CTSL in apoptosis in HCC, mice with pancreatic islet cancer with a knockout in CTSL demonstrated a 337% increase in apoptosis [90][29]. In line, the injection of lung cancer cells with decreased CTSL expression in mice showed increased apoptosis in tumors compared to mice injected with tumor cells with normal CTSL expression [91][30]. Hence, the role of CTSL seems to be similar to CTSS, i.e., to inhibit apoptosis.
Altogether, cathepsins play a key role in modulating apoptosis in multiple types of cancer. The specific type of cathepsin and possibly the type of cancer determines whether this role is pro- or anti-apoptotic.
2.3. Cathepsins Induce Angiogenesis in HCC
Angiogenesis is the process whereby new blood vessels form from existing vessels. This process occurs in physiological conditions via tightly regulated mechanisms [92][31]. Whereas, in tumors, this process is severely deregulated. Tumors rely on new blood vessels for continuous growth and tumor cells can self-induce angiogenesis (angiogenic switch) by secreting pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF).
HCC is characterized by hypervascularity with vascular structural and functional abnormalities [93,94][32][33]. This importance of angiogenesis in HCC is also reflected by the fact that most approved treatments for advanced HCC patients, including sorafenib, target several angiogenic pathways, including the pro-angiogenic VEGF/VEGF receptor (VEGFR) pathway [40,93][32][34]. In addition, the serum levels of VEGF have been shown to be increased along the progression of HCC. Furthermore, HCC patients with high serum levels of VEGF also showed a significant lower overall survival compared to patients with low serum levels of VEGF (±8 vs. 38 months [95][35]) [96][36]. In addition, the pro-angiogenic bFGF was found to be increased in HCC and a better biomarker for HCC diagnosis compared to the standard marker AFP (based on AUROC curves) [95][35].
In HCC, predominantly CTSB and CTSS have been demonstrated to be positively associated with angiogenesis in HCC (Figure 2) [97][37]. Cathepsins seem to be specifically involved in the degradation of the extracellular matrix (ECM), an essential process to allow new vessel sprouts to arise. Several in vitro studies demonstrated the effect of cathepsin inhibition on angiogenesis. Upon CTSB knockdown in HCC (Huh-7 cells transfected with hepatitis B spliced protein) cells, specific proteins involved in angiogenesis (matrix metalloprotease, MMP; and urokinase-type plasminogen activator, uPA) were reduced in gene expression. In addition, when the medium of these cells treated with siRNA for CTSB was transferred to endothelial cells (HMEC-1 cells), they observed reduced endothelial capillary formation compared to a control [98][38]. In addition to CTSB, CTSS has also been demonstrated to be involved in angiogenesis in HCC. Upon knockdown of CTSS in HCC cells (MHCC-97-H cells), reduced secretion of pro-angiogenic factor VEGF was observed. Furthermore, endothelial (HUVEC) cells showed reduced tube formation after being treated with medium obtained from the siRNA-treated HCC cells [73,99][11][39]. In line, CTSS overexpression in endothelial (HUVEC) cells increased tube formation compared to the control [99][39].
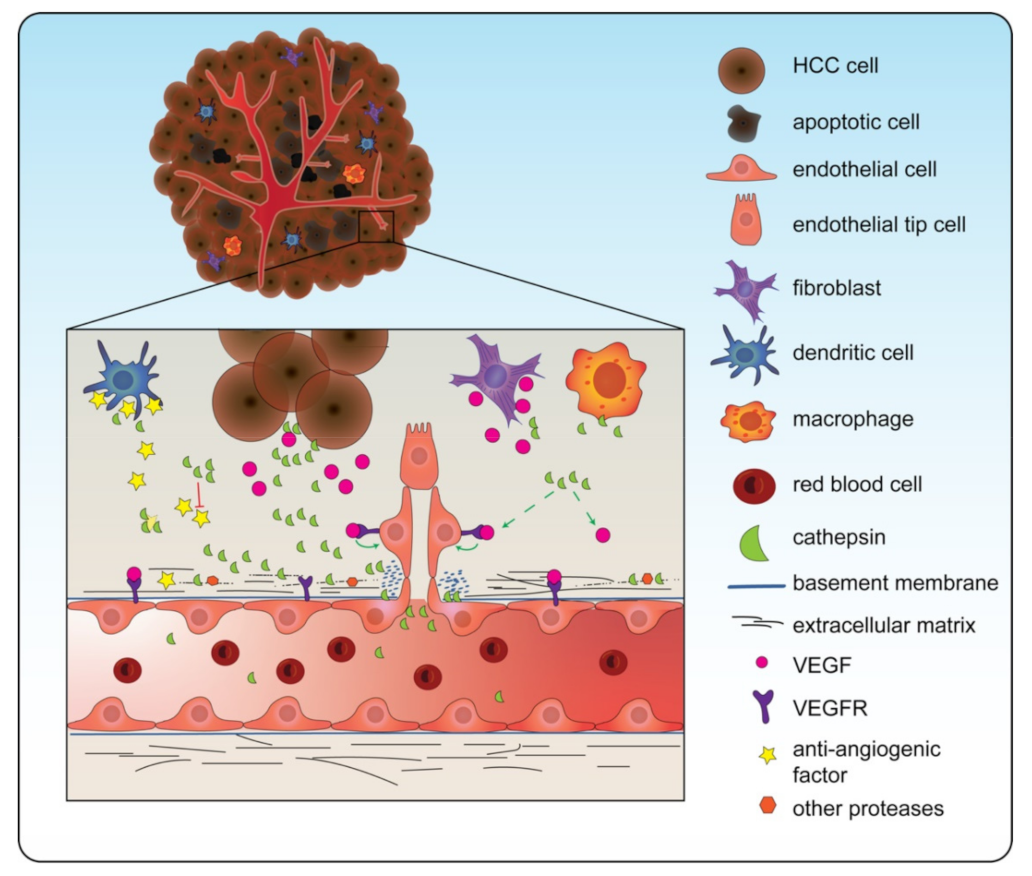
Figure 2. Cathepsins are involved in angiogenesis. Cathepsins are secreted into the tumor microenvironment (TME) by cancer cells but also by other cells from the TME such as fibroblasts and macrophages. These extracellular cathepsins increase the levels of vascular endothelial growth factor (VEGF), which induces angiogenesis. Furthermore, they cleave the extracellular matrix components, making it possible for the growing tip to develop. In line, cathepsins cleave anti-angiogenic factors. Note: in this figure CTSD, CTSB, CTSS, and CTSL are all depicted as one green symbol for ease of concept.
Other cathepsins might also play an important role in the angiogenesis process. Although not directly demonstrated for HCC, studies using other types of cancer demonstrated a role for both CTSL and CTSD in angiogenesis.
In a co-culture of CTSL-knocked-down gastric cancer cells with endothelial cells, a reduction in tubule formation was observed whereas CTSL overexpression led to increased tubule formation. Furthermore, CTSL knockdown in vivo through the use of the chorioallantoic membrane (CAM) model decreased the number of blood vessels around the gastric cancer tumor compared to the control [100,101][40][41]. Taking into account the observed overexpression of CTSL in HCC and its function in ECM degradation, these data in gastric cancer suggest that CTSL also stimulates angiogenesis in HCC. In line, mice xenografted with breast cancer cells that were overexpressed in CTSD demonstrated an increase in the number of tumor micro-vessels compared to mice that were xenografted with cells with normal CTSD expression [102][42]. Similar to CTSL, CTSD is overexpressed in HCC and can cleave the ECM. Therefore, it is suggested that CTSD also plays an important role in HCC angiogenesis.
2.4. Cathepsins Play a Crucial Role in Activating Invasion and Metastasis in HCC
Most cancer-related deaths occur due to infestations of the cancer cells into different body parts, or metastases. Cancer cells can leave their primary tumor location through the blood vessels to invade distant body sites. By invading, the malignant cells have to cross the basal membrane and the interstitial connective tissue [18][8]. Proteases play an essential role in these processes [103][43].
The role of cathepsins in the process of invasion and metastasis is illustrated in Figure 3 [104,105][44][45]. They are involved in three stages of tumor invasion: (1) cathepsins that migrate to the cell membrane have the ability to activate other ECM-degrading proteases, such as MMPs and uPAs; (2) cathepsins that are secreted by cancer or by immune cells in the TME can cleave (together with other proteases) the ECM or the basement membrane; and (3) cathepsins can break the interaction between cells, by cleaving the adherens junction components, such as E-cadherin [105][45]. Cathepsins can cleave these targets of tumor invasion upon attachment to the cancer cell membrane or they can be secreted into the extracellular milieu. CTSB, for example, is often found at the cell membrane in malignant cells and at the edge of the invasive tumor [103,106,107][43][46][47].
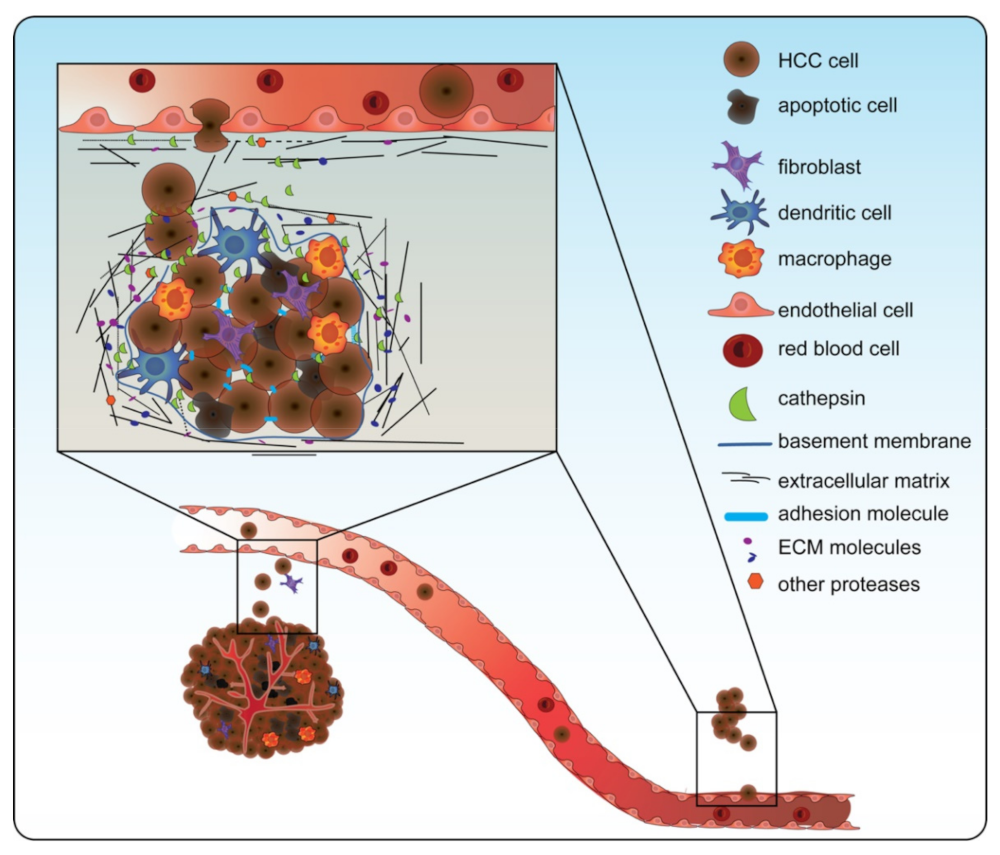
Figure 3. Cathepsins are involved in tumor invasion and metastasis. Cathepsins are involved in tumor invasion and metastasis by (1) going to the surface of the cancer cell or being secreted to activate other proteases such as matrix metalloproteases (MMPs) and urokinase-type plasminogen activator (uPAs); (2) cleaving the extracellular matrix (ECM) together with the other proteases; and (3) breaking the junctions between cells. In doing so, cathepsins contribute to the epithelial-to-mesenchymal transition, aiding in invasion through the ECM and entry into the blood vessel. Note: in this figure, CTSD, CTSB, CTSS, and CTSL are all depicted as one green symbol for ease of concept.
In HCC, CTSB induced the invasion of a cohesive multicellular group of cells (collective invasion) in a 3D invasion in vitro (HepG2 and Huh-7 cells) model. In this restudyearch, the expression of CTSB was enhanced through CD147, a transmembrane protein that is thought to be involved in invasion and metastasis [108][48]. The role of CTSB in invasion and migration was further established in wound healing and transwell invasion assays (in Huh-7 cells). In addition, CTSB overexpression in HCC cells with low metastatic potential (MHCC-97-L cells) demonstrated increased migration and invasion. In line, when CTSB was knocked down in an HCC cell line with high metastatic potential (MHCC-97-H), these characteristics diminished [11][49]. Furthermore, CTSB knockdown led to reduced expression of important proteases: MMP9 and uPA [11,98][38][49]. Along the same line, evidence points towards the involvement of CTSS in invasion and metastasis. CTSS is barely expressed in normal hepatocytes but significantly expressed in HCC hepatocytes [43][50]. Knockdown of CTSS in HCC (MHCC-97-H) cells with high metastatic potential significantly decreased invasion in a transwell invasion assay and motility in a wound healing assay [73][11]. Moreover, an increased CTSS expression level has been shown to be positively correlated with extrahepatic metastasis in HCC patients [43][50].
While the role of CTSD and CTSL has not been specifically demonstrated in HCC, CTSD has been shown to be directly involved in invasion and metastasis in various other cancer types, including breast cancer. CTSD overexpression in breast cancer (MCF-7 and MDA-MB-231) cells promoted migration and invasion, and in addition, it reduced the protein levels of epithelial markers, such as E-cadherin. Similarly, in this restudyearch, CTSD overexpression in vivo increased the number of metastases [109][51]. In line, a role for CTSL in invasion and metastasis was demonstrated in ovarian carcinoma cells. Overexpression of CTSL in these cells enhanced their ability to invade and metastasize, whereas downregulation of CTSL decreased these abilities [110][52]. Considering the role of CTSD and CTSL in other cancer types and their overexpression in HCC, these data suggest that these cathepsins also stimulate invasion and metastasis in the context of HCC.
2.5. Cathepsins Take Part in the Reprograming of Energy Metabolism in HCC
The energy metabolism in cancer cells is frequently deregulated to sustain their increased energy requirements for continuous growth and proliferation. An example of deregulated energy metabolism is the “Warburg effect”, which entails that cancer cells prefer glycolysis over oxidative phosphorylation [17][7]. It became clear recently that “metabolic reprogramming” is imperative for the cancer to prevail and progress. Several metabolic pathways, such as the glutaminolysis pathway that catabolizes glutamine into the metabolites glutamate or α-ketoglutarate, are increased in cancer [111][53]. Furthermore, several metabolic factors, such as insulin-like growth factor 1 (IGF-1), can influence growth and survival pathways, e.g., the PI3K/Akt/mTOR pathway [112][54].
Cathepsins play a role in this metabolic deregulation through IGF-1 signaling. IGF-1 has been shown to significantly promote cell proliferation, migration, and invasion in HCC (Hepa1-6 and H22) cell lines [113][55]. This pro-cancerous effect was exerted through CTSB, as upon CTSB depletion, the IGF-1 stimulatory effects were reversed. Furthermore, when WT mice and diabetic (KK-ay; with high IGF-1 levels) mice were xenografted with HCC (Hepa1-6) cancer cells, it was observed that the cancer expanded faster in the diabetic mice (i.e., by, among others, the high levels of IGF-1). Furthermore, upon knockdown of CTSB, the diabetic mice showed a significant reduction in tumor expansion compared to the diabetic mice that had normal CTSB levels, indicating that CTSB was the effector protein of IGF-1 [113][55]. Similarly, upon CTSL knockdown, less proliferation and migration were observed in vitro upon stimulation with IGF-1 compared to cells that had normal CTSL expression [91][30].
Not only IGF-1 is an upstream factor of cathepsin activity. Additionally, the addition of glucose led to increased levels of cathepsins in vitro. Sung et al. demonstrated that the addition of glucose to the medium of human peritoneal mesothelial cells led to an increase in the CTSB levels [114][56]. Since glucose metabolism is upregulated in HCC [115][57], this points towards a potential link between deregulated metabolism and cathepsin activity.
2.6. Cathepsins Promote Tumor Immune Evasion
Immune surveillance is a process whereby immune cells monitor to detect and eliminate cancer cells. Cancer cells have, however, developed a mechanism to avoid this immune destruction [116][58]. Malignant cells can secrete immunosuppressive factors such as TGF-β, which recruits regulatory T cells, myeloid-derived suppressor cells (MDSCs), and other immunosuppressive cells, leading to the suppression of the immune response [17][7].
Cathepsins can be expressed and secreted by immune cells and they have physiological functions in the immune system [117][59]. Cathepsins have been found to play a part in toll-like receptor signaling and cytokine activation/inhibition [118,119][60][61]. In addition, cathepsins, mainly CTSS, play an important role in the processing and presenting of antigens in the antigen-presenting cells, e.g., macrophages and dendritic cells [120][62].
Cathepsins not only have a physiological function in the immune system but are also key players in the evasion of the immune system present in cancers. The TME is a significant contributor to the suppressed immune response that is present in HCC [121][63]. Among the cells present in the TME, including in HCC, tumor-associated macrophages (TAMs) are key players. TAMs have an M2 macrophage-like anti-inflammatory phenotype, unlike the M1 macrophage phenotype, which is proinflammatory [122][64]. Because TAMs suppress the immune response, they are tumor-promoting; hence, their presence is generally related with poor prognosis [123][65]. TAMs secrete CTSB, CTSL, and CTSS [124,125][66][67] and therefore, TAMs also provoke tumor growth, angiogenesis, invasion, and metastasis responses. Inhibition of these cathepsins in vitro (human M2 polarized macrophages derived from PBMCs used as a representative cell of TAM) led to an increase in the expression of proinflammatory cytokines such as TNF-α and IL-1β and also to increased protein levels of NOS2 (M1 marker), among others [125][67]. As such, these data suggest that CTSB, CTSL, and CTSS inhibition in M2 macrophages drives a shift to M1 polarization [125,126][67][68]. Hence, the expression of CTSB, CTSL, and CTSS represents an important way for TAMs to evade the immune system. In line, other myeloid cells in the TME have been shown to gain an M2 phenotype, mainly through CTSS. These M2 phenotype cells favor the immunosuppressive regulatory T cells over cytotoxic CD8+ T cells, contributing even further to the immunosuppressive environment [127][69]. In addition, the TME contains MDSCs, which also harbor an immunosuppressive function. Studies found that the expression of CTSB was increased in MDSCs, thereby contributing to HCC progression as well [128][70].
The levels of SIRT1 in HCC cells are high and seem to have a pro-tumor role by inducing an immunosuppressive phenotype, among others [131,132,133,134][71][72][73][74].
Hence, these data suggest that, in general, cathepsins play an active role in the deregulated immune system surrounding HCC.
2.7. Genomic Variation in Cathepsin Genes Can Alter Tumor Development
Genetic alternations are the driving force behind cancer development. Several single nucleotide polymorphisms (SNPs) in the genes that encode cathepsins have been associated with cancer development. Chen et al. demonstrated that in an Asian population, an SNP present in the CTSB gene (rs13332) was significantly associated with the risk of HCC (with an adjusted odds ratio of 2.2) [135][75]. In addition, Cui et al. showed that the genotype and allele distribution of a CTSB SNP (rs12898) between HCC patients and healthy controls was significantly different [136][76].
For CTSL and CTSD, there is no direct evidence available for the effect of genetic variations in HCC or cancer in general. CTSS, however, is often mutated in follicular lymphoma. It was determined that a specific variation (Y132D) increases CTSS activity, which has tumor-promoting effects [137][77].
These data indicate that variations in the cathepsin genes can promote cancer and that more research should be carried out to study the effect of the various existing variants.
References
- Willstätter, R.; Bamann, E. Über die Proteasen der Magenschleimhaut. Erste Abhandlung Über die Enzyme der Leukocyten; Hoppe-Seyler’s Zeitschrift Fur Physiologische Chemie; De Gruyter: Berlin, Germany, 1929; Volume 180, pp. 127–143.
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431.
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1679.
- Turk, B.; Turk, D.; Turk, V. Lysosomal cysteine proteases: More than scavengers. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1477, 98–111.
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 68–88.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Ruan, J.; Zheng, H.; Fu, W.; Zhao, P.; Su, N.; Luo, R. Increased expression of cathepsin L: A novel independent prognostic marker of worse outcome in hepatocellular carcinoma patients. PLoS ONE 2014, 9, e112136.
- Xu, Z.Z.; Xiu, P.; Lv, J.W.; Wang, F.H.; Dong, X.F.; Liu, F.; Li, T.; Li, J. Integrin αvβ3 is required for cathepsin B-induced hepatocellular carcinoma progression. Mol. Med. Rep. 2015, 11, 3499–3504.
- Fan, Q.; Wang, X.; Zhang, H.; Li, C.; Fan, J.; Xu, J. Silencing cathepsin S gene expression inhibits growth, invasion and angiogenesis of human hepatocellular carcinoma in vitro. Biochem. Biophys. Res. Commun. 2012, 425, 703–710.
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect Biol. 2012, 4, a011189.
- Peng, S.; Yang, Q.; Li, H.; Pan, Y.; Wang, J.; Hu, P.; Zhang, N. CTSB knockdown inhibits proliferation and tumorigenesis in HL-60 cells. Int. J. Med. Sci. 2021, 18, 1484–1491.
- Ketterer, S.; Mitschke, J.; Ketscher, A.; Schlimpert, M.; Reichardt, W.; Baeuerle, N.; Hess, M.E.; Metzger, P.; Boerries, M.; Peters, C.; et al. Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling. Nat. Commun. 2020, 11, 5133.
- Zhang, L.; Wang, H.; Xu, J.; Zhu, J.; Ding, K. Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicol. Lett. 2014, 228, 248–259.
- Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. cFLIPL prevents TRAIL-induced apoptosis of hepatocellular carcinoma cells by inhibiting the lysosomal pathway of apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1337–G1346.
- Caruso, J.A.; Mathieu, P.A.; Joiakim, A.; Zhang, H.; Reiners, J.J., Jr. Aryl hydrocarbon receptor modulation of tumor necrosis factor-alpha-induced apoptosis and lysosomal disruption in a hepatoma model that is caspase-8-independent. J. Biol. Chem. 2006, 281, 10954–10967.
- Ullio, C.; Casas, J.; Brunk, U.T.; Sala, G.; Fabriàs, G.; Ghidoni, R.; Bonelli, G.; Baccino, F.M.; Autelli, R. Sphingosine mediates TNFα-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J. Lipid Res. 2012, 53, 1134–1143.
- Desideri, E.; Ciriolo, M.R. Inhibition of JNK increases the sensitivity of hepatocellular carcinoma cells to lysosomotropic drugs via LAMP2A destabilization. Cell Death Discov. 2021, 7, 29.
- Ruiz-Blázquez, P.; Pistorio, V.; Fernández-Fernández, M.; Moles, A. The multifaceted role of cathepsins in liver disease. J. Hepatol. 2021, 75, 1192–1202.
- Droga-Mazovec, G.; Bojič, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of Bid and antiapoptotic Bcl-2 homologues*. J. Biol. Chem. 2008, 283, 19140–19150.
- de Castro, M.A.; Bunt, G.; Wouters, F.S. Cathepsin B launches an apoptotic exit effort upon cell death-associated disruption of lysosomes. Cell Death Discov. 2016, 2, 16012.
- Gan, Y.; Zhao, X.; Hu, J.; Wang, Z.G.; Zhao, X.T. HCCS1 overexpression induces apoptosis via cathepsin D and intracellular calcium, and HCCS1 disruption in mice causes placental abnormality. Cell Death Differ. 2008, 15, 1481–1490.
- Roberts, L.R.; Kurosawa, H.; Bronk, S.F.; Fesmier, P.J.; Agellon, L.B.; Leung, W.Y.; Mao, F.; Gores, G.J. Cathepsin B contributes to bile salt-induced apoptosis of rat hepatocytes. Gastroenterology 1997, 113, 1714–1726.
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137.
- Wang, X.; Xiong, L.; Yu, G.; Li, D.; Peng, T.; Luo, D.; Xu, J. Cathepsin S silencing induces apoptosis of human hepatocellular carcinoma cells. Am. J. Transl. Res. 2015, 7, 100–110.
- Seo, S.U.; Min, K.-j.; Woo, S.M.; Kwon, T.K. Z-FL-COCHO, a cathepsin S inhibitor, enhances oxaliplatin-mediated apoptosis through the induction of endoplasmic reticulum stress. Exp. Mol. Med. 2018, 50, 1–11.
- Fei, M.; Zhang, L.; Wang, H.; Zhu, Y.; Niu, W.; Tang, T.; Han, Y. Inhibition of cathepsin S induces mitochondrial apoptosis in glioblastoma cell lines through mitochondrial stress and autophagosome accumulation. Front. Oncol. 2020, 10, 516746.
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556.
- Navab, R.; Pedraza, C.; Fallavollita, L.; Wang, N.; Chevet, E.; Auguste, P.; Jenna, S.; You, Z.; Bikfalvi, A.; Hu, J.; et al. Loss of responsiveness to IGF-I in cells with reduced cathepsin L expression levels. Oncogene 2008, 27, 4973–4985.
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257.
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The role of angiogenesis in hepatocellular carcinoma. Clin. Cancer Res. 2019, 25, 912–920.
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301.
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616.
- Joo, Y.Y.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Yoon, S.K. Circulating pro- and anti-angiogenic factors in multi-stage liver disease and hepatocellular carcinoma progression. Sci. Rep. 2019, 9, 9137.
- Zhan, P.; Qian, Q.; Yu, L.K. Serum VEGF level is associated with the outcome of patients with hepatocellular carcinoma: A meta-analysis. Hepatobiliary Surg. Nutr. 2013, 2, 209–215.
- Joyce, J.A.; Baruch, A.; Chehade, K.; Meyer-Morse, N.; Giraudo, E.; Tsai, F.-Y.; Greenbaum, D.C.; Hager, J.H.; Bogyo, M.; Hanahan, D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell 2004, 5, 443–453.
- Chen, W.N.; Chen, J.Y.; Jiao, B.Y.; Lin, W.S.; Wu, Y.L.; Liu, L.L.; Lin, X. Interaction of the hepatitis B spliced protein with cathepsin B promotes hepatoma cell migration and invasion. J. Virol. 2012, 86, 13533–13541.
- Zhang, Z.; Zhang, H.; Peng, T.; Li, D.; Xu, J. Melittin suppresses cathepsin S-induced invasion and angiogenesis via blocking of the VEGF-A/VEGFR-2/MEK1/ERK1/2 pathway in human hepatocellular carcinoma. Oncol. Lett. 2016, 11, 610–618.
- Pan, T.; Jin, Z.; Yu, Z.; Wu, X.; Chang, X.; Fan, Z.; Li, F.; Wang, X.; Li, Z.; Zhou, Q.; et al. Cathepsin L promotes angiogenesis by regulating the CDP/Cux/VEGF-D pathway in human gastric cancer. Gastric Cancer 2020, 23, 974–987.
- Rebbaa, A.; Chu, F.; Sudha, T.; Gallati, C.; Dier, U.; Dyskin, E.; Yalcin, M.; Bianchini, C.; Shaker, O.; Mousa, S.A. The anti-angiogenic activity of NSITC, a specific cathepsin L inhibitor. Anticancer Res. 2009, 29, 4473–4481.
- Berchem, G.; Glondu, M.; Gleizes, M.; Brouillet, J.P.; Vignon, F.; Garcia, M.; Liaudet-Coopman, E. Cathepsin-D affects multiple tumor progression steps in vivo: Proliferation, angiogenesis and apoptosis. Oncogene 2002, 21, 5951–5955.
- Koblinski, J.E.; Ahram, M.; Sloane, B.F. Unraveling the role of proteases in cancer. Clin. Chim. Acta 2000, 291, 113–135.
- Poole, A.R.; Tiltman, K.J.; Recklies, A.D.; Stoker, T.A. Differences in secretion of the proteinase cathepsin B at the edges of human breast carcinomas and fibroadenomas. Nature 1978, 273, 545–547.
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64.
- Duffy, M.J. The role of proteolytic enzymes in cancer invasion and metastasis. Clin. Exp. Metastasis 1992, 10, 145–155.
- Tan, G.J.; Peng, Z.K.; Lu, J.P.; Tang, F.Q. Cathepsins mediate tumor metastasis. World J. Biol. Chem. 2013, 4, 91–101.
- Wang, S.J.; Chao, D.; Wei, W.; Nan, G.; Li, J.Y.; Liu, F.L.; Li, L.; Jiang, J.L.; Cui, H.Y.; Chen, Z.N. CD147 promotes collective invasion through cathepsin B in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 145.
- Ruan, J.; Zheng, H.; Rong, X.; Rong, X.; Zhang, J.; Fang, W.; Zhao, P.; Luo, R. Over-expression of cathepsin B in hepatocellular carcinomas predicts poor prognosis of HCC patients. Mol. Cancer 2016, 15, 17.
- Xu, J.; Li, D.; Ke, Z.; Liu, R.; Maubach, G.; Zhuo, L. Cathepsin S is aberrantly overexpressed in human hepatocellular carcinoma. Mol. Med. Rep. 2009, 2, 713–718.
- Zhang, C.; Zhang, M.; Song, S. Cathepsin D enhances breast cancer invasion and metastasis through promoting hepsin ubiquitin-proteasome degradation. Cancer Lett. 2018, 438, 105–115.
- Zhang, W.; Wang, S.; Wang, Q.; Yang, Z.; Pan, Z.; Li, L. Overexpression of cysteine cathepsin L is a marker of invasion and metastasis in ovarian cancer. Oncol. Rep. 2014, 31, 1334–1342.
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680.
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336.
- Lei, T.; Ling, X. IGF-1 promotes the growth and metastasis of hepatocellular carcinoma via the inhibition of proteasome-mediated cathepsin B degradation. World J. Gastroenterol. 2015, 21, 10137–10149.
- Sung, S.A.; Kim, D.H.; Oh, K.H.; Han, S.Y.; Han, K.H. The role of cathepsin B in peritoneal fibrosis due to peritoneal dialysis. Int. J. Nephrol. 2019, 2019, 4150656.
- Shang, R.Z.; Qu, S.B.; Wang, D.S. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J. Gastroenterol. 2016, 22, 9933–9943.
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312.
- Conus, S.; Simon, H.U. Cathepsins and their involvement in immune responses. Swiss Med. Wkly. 2010, 140, w13042.
- Matsumoto, F.; Saitoh, S.-i.; Fukui, R.; Kobayashi, T.; Tanimura, N.; Konno, K.; Kusumoto, Y.; Akashi-Takamura, S.; Miyake, K. Cathepsins are required for Toll-like receptor 9 responses. Biochem. Biophys. Res. Commun. 2008, 367, 693–699.
- Flynn, C.M.; Garbers, Y.; Düsterhöft, S.; Wichert, R.; Lokau, J.; Lehmann, C.H.K.; Dudziak, D.; Schröder, B.; Becker-Pauly, C.; Rose-John, S.; et al. Cathepsin S provokes interleukin-6 (IL-6) trans-signaling through cleavage of the IL-6 receptor in vitro. Sci. Rep. 2020, 10, 21612.
- Shi, G.P.; Villadangos, J.A.; Dranoff, G.; Small, C.; Gu, L.; Haley, K.J.; Riese, R.; Ploegh, H.L.; Chapman, H.A. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity 1999, 10, 197–206.
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-associated macrophages in hepatocellular carcinoma pathogenesis, prognosis and therapy. Cancers 2022, 14, 226.
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429.
- Zhang, Q.-w.; Liu, L.; Gong, C.-y.; Shi, H.-s.; Zeng, Y.-h.; Wang, X.-z.; Zhao, Y.-w.; Wei, Y.-q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946.
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255.
- Oelschlaegel, D.; Weiss Sadan, T.; Salpeter, S.; Krug, S.; Blum, G.; Schmitz, W.; Schulze, A.; Michl, P. Cathepsin inhibition modulates metabolism and polarization of tumor-associated macrophages. Cancers 2020, 12, 2579.
- Yang, M.; Liu, J.; Shao, J.; Qin, Y.; Ji, Q.; Zhang, X.; Du, J. Cathepsin S-mediated autophagic flux in tumor-associated macrophages accelerate tumor development by promoting M2 polarization. Mol. Cancer 2014, 13, 43.
- Fuchs, N.; Meta, M.; Schuppan, D.; Nuhn, L.; Schirmeister, T. Novel opportunities for cathepsin S inhibitors in cancer immunotherapy by nanocarrier-mediated delivery. Cells 2020, 9, 2021.
- Boutté, A.M.; McDonald, W.H.; Shyr, Y.; Yang, L.; Lin, P.C. Characterization of the MDSC proteome associated with metastatic murine mammary tumors using label-free mass spectrometry and shotgun proteomics. PLoS ONE 2011, 6, e22446.
- Chen, H.C.; Jeng, Y.M.; Yuan, R.H.; Hsu, H.C.; Chen, Y.L. SIRT1 promotes tumorigenesis and resistance to chemotherapy in hepatocellular carcinoma and its expression predicts poor prognosis. Ann. Surg. Oncol. 2012, 19, 2011–2019.
- Farcas, M.; Gavrea, A.A.; Gulei, D.; Ionescu, C.; Irimie, A.; Catana, C.S.; Berindan-Neagoe, I. SIRT1 in the development and treatment of hepatocellular carcinoma. Front. Nutr. 2019, 6, 148.
- Molla, M.D.; Dessie, G.; Akalu, Y.; Ayelign, B. Hepatocellular expression of SIRT1 and its effect on hepatocellular carcinoma progression: A future therapeutic perspective. Int. J. Hepatol. 2020, 2020, 2374615.
- Choi, H.N.; Bae, J.S.; Jamiyandorj, U.; Noh, S.J.; Park, H.S.; Jang, K.Y.; Chung, M.J.; Kang, M.J.; Lee, D.G.; Moon, W.S. Expression and role of SIRT1 in hepatocellular carcinoma. Oncol. Rep. 2011, 26, 503–510.
- Chen, T.P.; Yang, S.F.; Lin, C.W.; Lee, H.L.; Tsai, C.M.; Weng, C.J. A4383C and C76G SNP in Cathepsin B is respectively associated with the high risk and tumor size of hepatocarcinoma. Tumour. Biol. 2014, 35, 11193–11198.
- Cui, M.; Chen, Q.; He, C.; Wang, N.; Yu, Y.; Sun, Z.; Lin, Z.; Cui, H.; Jin, S.; Park, J.Y. A single nucleotide polymorphism CTSB rs12898 is associated with primary hepatic cancer in a Chinese population. Int. J. Clin. Exp. Pathol. 2019, 12, 3063–3069.
- Bararia, D.; Hildebrand, J.A.; Stolz, S.; Haebe, S.; Alig, S.; Trevisani, C.P.; Osorio-Barrios, F.; Bartoschek, M.D.; Mentz, M.; Pastore, A.; et al. Cathepsin S alterations induce a tumor-promoting immune microenvironment in follicular lymphoma. Cell Rep. 2020, 31, 107522.
More