Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Duncan Smith and Version 2 by Rita Xu.
β-Thalassemia is one of the most common genetically inherited disorders worldwide, and it is characterized by defective β-globin chain synthesis leading to reduced or absent β-globin chains. The excess α-globin chains are the key factor leading to the death of differentiating erythroblasts in a process termed ineffective erythropoiesis, leading to anemia and associated complications in patients. The mechanism of ineffective erythropoiesis in β-thalassemia is complex and not fully understood. Autophagy is primarily known as a cell recycling mechanism in which old or dysfunctional proteins and organelles are digested to allow recycling of constituent elements.
- autophagy
- apoptosis
- ER stress
- β-thalassemia
1. Introduction
β-Thalassemia, one of the most common genetic inheritance disorders worldwide, is caused by the absence or reduced presence of β-globin chains in hemoglobin (Hb) molecules [1]. The Hb-producing cells accumulate excess α-globin, which forms cytotoxic intracellular precipitates that impair erythroid cell production in a process known as ineffective erythropoiesis, a major determinant of β-thalassemia pathophysiology [2]. Erythrokinetic and ferrokinetic studies in β-thalassemia patients indicated that about 65% of erythroblasts die before becoming mature red blood cells (RBCs) [3]. Autophagy plays an important role in organelle clearance during terminal erythroid differentiation of erythrocytes [4], in addition to its cellular role of recycling macromolecules in cells to maintain homeostasis [5]. Autophagy can be induced by extracellular or intracellular stresses such as nutrient and growth factor deprivation [6], hypoxia [7], calcium (Ca2+) dysregulation [8], endoplasmic reticulum (ER) stress [9], and oxidative stress [10].
2. Clinical Manifestation of β-Thalassemia and β-Thalassemia/HbE Diseases
2.1. Etiology and Epidemiology of β-Thalassemia and β-Thalassemia/HbE Diseases
The Hb tetramer consists of two different pairs of globin chains and heme, such as adult Hb or HbA which consists of two α-globin and two β-globin chains (α2β2) together with four heme-iron complexes. It is Hb that provides RBCs with the capacity to carry oxygen molecules to tissues and to remove carbon dioxide. Hemoglobinopathy is characterized by a reduction in globin production (called thalassemia) or by the presence of an abnormal Hb, such as HbS (Sickle cell hemoglobin; α2βS2), which results from a single base mutation of the β-globin gene at codon 6, leading to an amino acid change from glutamine to valine [11]. Additionally, HbE (α2βE2), the second most prevalent abnormal Hb of the world, is both a thalassemia and an abnormal Hb. In HbE, there is a single base mutation of β-globin gene at codon 26 leading to the amino acid change from glutamine to lysine, which additionally creates an aberrant splicing of the βE-globin pre-mRNA leading to a reduction of βE-globin chains [12].
Hemoglobinopathy is among the most common genetic disorders worldwide, and can be found in the Mediterranean region, the Middle East, the Indian subcontinent, and throughout Southeast Asia in a line stretching from southern China down the Malaysian peninsula to the Indonesian islands. Hemoglobinopathy is widely distributed, and approximately 5.2% of the world’s population carry significant globin variants including HbS, HbC, HbE, HbD, β-thalassemia, and α-thalassemia (Figure 1) [13]. Therefore, the worldwide birth rate of newborns who are homozygous or compound heterozygous for symptomatic thalassemia, including HbBart’s hydrops fetalis (homozygous α0-thalassemia), HbH disease (α0/α+-thalassemia), β-thalassemia major (homozygous β0-thalassemia), and β-thalassemia/HbE disease, is 0.46 per 1000 births. Although the most severe form of thalassemia is HbBart’s hydrops fetalis, the affected fetus dies in utero, and therefore the majority of severe thalassemia patients worldwide are homogygous β-thalassemia and β-thalassemia/HbE. The estimated number of births with β-thalassemia annually is approximately 40,618 of which 25,511 (62.8%) have severe anemia and require blood transfusions. However, only 11.7% of patients requiring blood transfusion can reached for treatment (Figure 1) [13]. In addition, HbE is common in Southeast Asia, Bangladesh, and Northeast India [14]. Increased global migration is a major factor that has increased the prevalence of hemoglobinopathies worldwide, leading to it becoming a global, rather than regional public health problem.
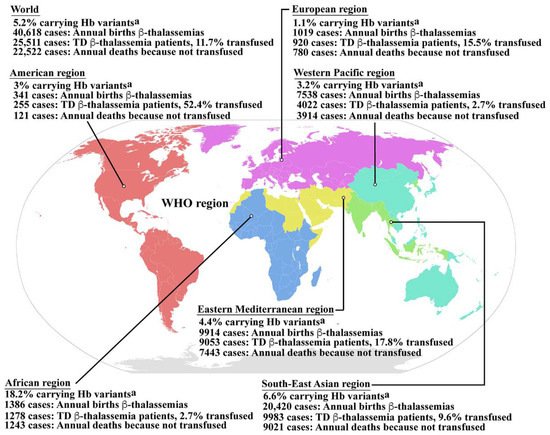
Figure 1. Epidemiology of β-thalassemia. Global epidemiological data including the percentages of populations carrying Hb variants, the number of newborns who have β-thalassemias per year, the number of patients who have TDT, and the percentage of TDT patients who are reached for blood transfusion, and the number of patients who died as a consequence of not receiving blood transfusion worldwide and in the individual sub-regions, according to the World Health Organization (WHO) as reported by Modell B and Darlison M published in the Bull World Health Organ 2008, 86, (6), 480–487 [13]. a Hemoglobin variants including HbS, HbC, HbE, HbD, etc., β-thalassemia and α-thalassemia. Hb; hemoglobin, TD β-thalassemia; transfusion dependent β-thalassemia. Source of WHO region map; wikipedia.org (accessed in September 2022). The map is in the public domain.
β-Thalassemia is a heterogeneous group of inherited disorders of Hb synthesis that is characterized by the absence or reduced presence of β-globin chains. Over 300 mutations in the β-globin gene that cause the β-thalassemia have been documented [15]. Clinical manifestations range from the β-thalassemia trait, which presents as a borderline asymptomatic anemia (β/β+ or β/β0); to β-thalassemia intermedia, alternatively termed non-transfusion dependent thalassemia (NTDT), which presents with mild-to-moderate symptoms as a result of a relative reduction in β-globin chain production (β+/β+ or β+/β0); to β-thalassemia major, alternatively termed transfusion dependent thalassemia (TDT), which results in severe symptoms as a consequence of the complete absence of β-globin chain synthesis (β0/β0) [2][16][2,16]. The compound heterozygosity caused by co-inheritance of β-thalassemia and HbE, which causes β-thalassemia/HbE disease (β0/βE or β+/βE), can result in a wide range of disease severity, with Hb levels ranging from 3 to 13 g/dL, resulting in mild or moderate (like β-thalassemia intermedia) to severe (like β-thalassemia major) presentation [2][17][2,17].
2.2. Pathophysiology of β-Thalassemia and β-Thalassemia/HbE Diseases
The β-thalassemia or β-thalassemia/HbE patients have imbalanced globin chains, with excess unbound α-globin chains in erythroblasts resulting in ineffective erythropoiesis (Figure 2). The patients suffer from chronic anemia that induces erythropoietin production and consequently stimulates massive erythropoiesis, extramedullary hematopoiesis, and increased iron absorption at the gastrointestinal tract [18]. Abnormal thalassemic RBCs are commonly cleared by macrophages in the sinusoids of spleen and by the reticuloendothelial (RE) system, where heme is degraded into a porphyrin ring and iron. The iron is recycled, while the porphyrin ring is oxidized to biliverdin and subsequently converted to unconjugated bilirubin and then passively taken into hepatocytes. High levels of bilirubin can cause jaundice and gallstones in patients. The increased number of damaged RBCs stimulate RE hyperplasia and can induce hepatosplenomegaly. In turn, the increased number of RBCs destroyed in the spleen can trigger hyperfunction of macrophages leading to hypersplenism, resulting in severe anemia. Splenectomy is a conventional treatment to cure hypersplenism, but it subsequently increases susceptibility to infection and can promote thrombosis. In addition, massive erythropoiesis in the bone marrow can induce bone changes that contribute to osteoporosis, while extramedullary hematopoiesis can interfere with organ functions leading to additional complications. The conventional treatment for patients with severe anemia is regular blood transfusions. However, increased gastrointestinal iron absorption together with multiple blood transfusion leads patients to suffer from iron overload, which is a serious complication that plays an important role in induced systemic abnormalities such as cardiovasculopathy, cirrhosis, diabetes mellitus, abnormal immunity, and other complications [19][20][21][22][23][24][19,20,21,22,23,24].
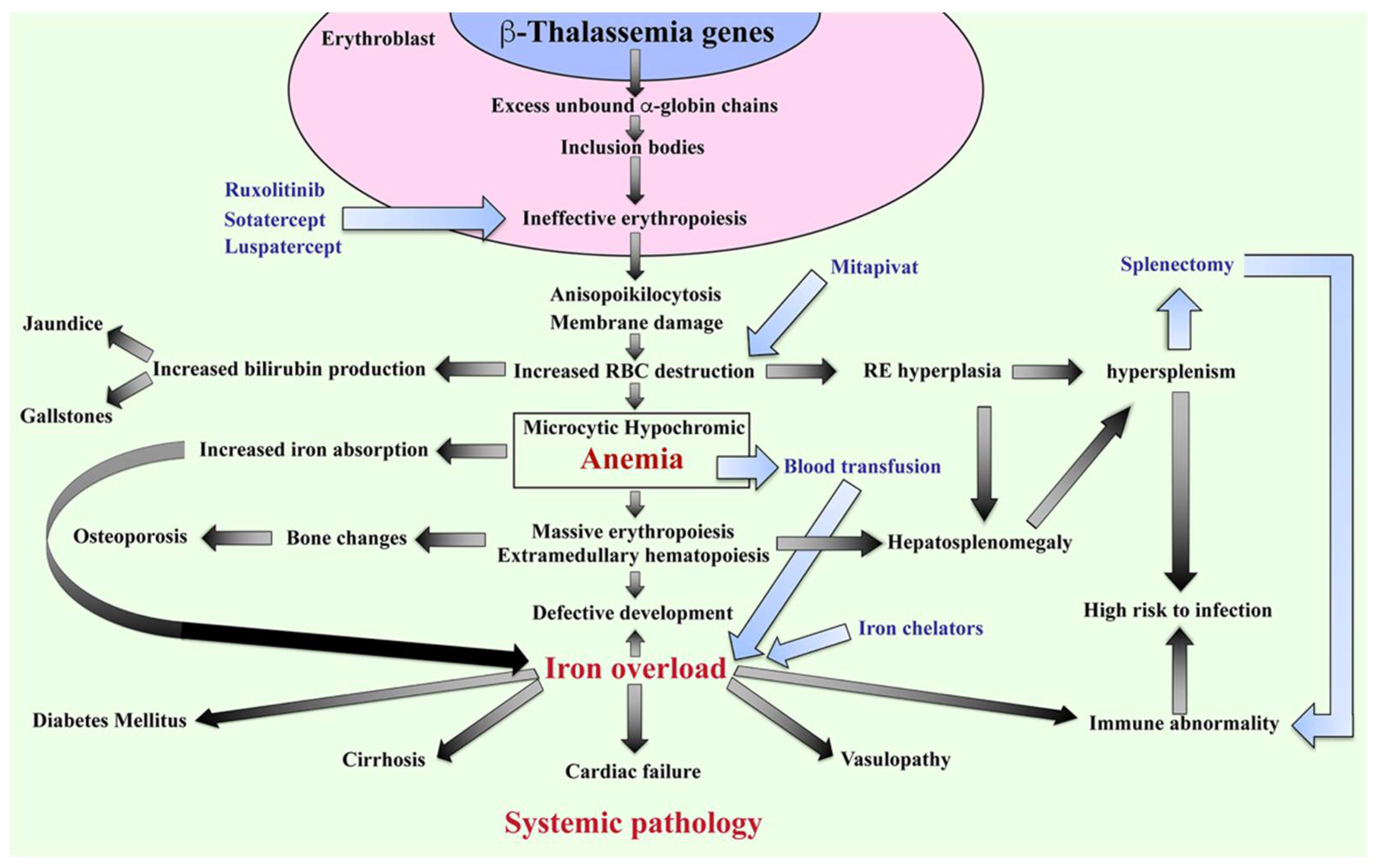
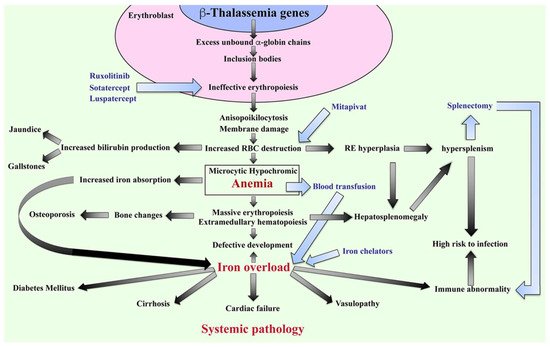
Figure 2. Pathophysiology of β-thalassemia. Defect in β-globin genes leads to reduced or absent β-globin production and consequent excess unbound α-globin chain accumulation and precipitation in erythroblasts, resulting in ineffective eythropoiesis. Abnormal RBCs, including anisopoikilocytosis, and RBCs with damaged membranes cause RBC destruction in the spleen leading to anemia, reticuloendothelial (RE) hyperplasia, and increased bilirubin production. Iron overload is a key factor in inducing a systemic pathology leading to increased mortality and morbidity in β-thalassemia patients. Blood transfusion and iron chelators are the conventional treatments for β-thalassemia. Curently, therapeutic drugs such as ruxolitinib, sotatercept, luspatercept, and mitapivat that target ineffective erythropoiesis by increasing Hb production are in clinical trials. Hb; hemoglobin, RBCs; red blood cells, and RE system; reticuloendothelial system.
2.3. Current Treatment for β-Thalassemia and β-Thalassemia/HbE Diseases
Currently, the recommended guidelines for medical management focus on blood transfusion and iron chelation to delay pathophysiological changes in patients. Therapeutic options such as stimulation of HbF induction using hydroxyurea, which aims to reduce the frequency of blood transfusion, are widely used. Curative treatments such as hematopoietic stem cell transplantation (HSCT) and gene therapy have been established but remain severely limited in application. Therefore, the clinical management for β-thalassemia depends on the age of the patients, the severity of anemia, and the individual responsiveness to treatments.
Blood transfusion: TDT patients who have anemia with Hb levels <7 g/dL require regular blood transfusions to maintain Hb levels of around 9–10 g/dL [25]. However, multiple blood transfusions cause iron overload and have a high risk of alloimmunization as well as an increased risk from blood-borne infectious diseases. Leukocyte-depleted RBCs are recommended to reduce the incidence of febrile and urticarial reactions as well as infectious cytomegalovirus contamination.
Iron chelation: Iron chelation therapy is recommended for patients who have serum ferritin levels of >1000 ng/mL to maintain serum ferritin levels of between 500–1000 ng/mL. The available iron chelators are deferoxamine, deferiprone and deferasirox. Deferoxamine is administered at a dose of 40–50 mg/kg by 8–12 h of subcutaneous infusion 5 days a week. Deferoxamine has the benefit of chelating iron from hepatocytes and cardiomyocytes; however, there is a high risk of Yersinia infections, hearing loss, retinopathy, poor growth, and allergy [26]. Deferiprone has oral tablet and syrup formulations, which are usually administered at 75–100 mg/kg/day. Deferiprone promotes the excretion of storage iron from parenchymal iron stores but has no advantage over deferoxamine in promoting reticuloendothelial iron excretion. The adverse effects of deferiprone are nausea, vomiting, abdominal pain, increased alanine aminotransferase levels, arthralgia, and neutropenia [26]. Combined therapy of deferoxamine and deferiprone has been recommended to treat patients with severe myocardial iron overload and for prevention and/or reversal of endocrinopathies. Deferasirox has dispersible tablet and film-coated tablet formulations that are administered once-daily at a dose of 20 or 30 mg/kg. Deferasirox is recommended for cardiac iron clearance, but even liver iron is lowered. The adverse effects of deferasirox are diarrhea, vomiting, nausea, abdominal pain, rash, increased alanine aminotransferase levels, and increased serum creatinine [26]. The iron chelator dosing and regimen for appropriate management of iron overload to avoid further organ toxicity and preserve organ function are the key concepts of treatment. Therefore, the optimal iron chelation therapy including the initiation time for treatment, close monitoring, and continuous adjustment needs careful consideration.
Stimulation of HbF production: Nowadays, hydroxyurea at a low dose (5–20 mg/kg/day for 5 days a week) is a therapeutic option for β-thalassemia, which aims to reduce the frequency of blood transfusions. However, hydroxyurea has potential severe adverse effects on neutropenia and thrombocytopenia that need careful monitoring. In addition, partial responders and non-responders to hydroxyurea treatment have been reported [27].
Stem cell transplantation: Allogenic HSCT could replace the ineffective endogenous erythropoiesis and can cure the disease. Hematopoietic stem cells (HSCs) can be harvested from peripheral blood and bone marrow of a human leukocyte antigen (HLA)-matched unrelated donor or HLA-haploidentical donor. HLA-identical sibling umbilical cord blood stem cell transplantation (UCBT) is a potential source of HSCs; however, it is limited to the yield of stem cells from a single unit of umbilical cord blood (UCB). Therefore, age and body weight of patient, ex vivo expansion of UCB stem cells, and enhanced homing to the bone marrow niche of UCB-derived HSCs are the key factors for good outcomes of UCBT. Moreover, patient age and disease status at time of transplantation as described by the Pesaro classification including the extent of hepatomegaly >2 cm, portal fibrosis, and history of inadequate iron chelation therapy are limitations. The risks of graft failure of stem cell transplantation are hyperplastic bone marrow, allosensitization due to multiple transfusion, graft-versus-host-disease (GvHD), infections, and iron overload [28].
Gene therapy: To avoid allogenic hematopoietic stem cell transplantation and failure because of GvHD, gene therapy is a therapeutic option that harvests hematopoietic stem cells from the patient and corrects the target gene in vitro and then transplants the cells back to the patient in a personalized medicine approach. The delivery system is the key factor of this approach to increase the efficiency of targeting the genomic DNA. Moreover, this approach is limited by the type of mutation, there is no universal gene therapy for all types of mutation to cure all types of thalassemia [29]. The limitation of curative treatments, for both hematopoietic stem cell transplantation and gene therapy is a consequence of the high risk and high cost, as well as a lack of suitable facilities and specialized physicians.
Therefore, understanding the mechanism of ineffective erythropoiesis as the major cause of anemia in β-thalassemia and β-thalassemia/HbE diseases could lead to novel therapeutics and management strategies to improve the quality and duration of the lives of patients.
3. Molecular Mechanism of Ineffective Erythropoiesis in β-Thalassemia
The ineffective erythropoiesis in β-thalassemia is characterized by a massive increase in erythroblasts proliferation, coupled with increased erythroblast cell death resulting in low levels of circulating RBCs causing chronic anemia. Studies on the bone marrow of β-thalassemia patients have found increased numbers of early erythroid precursor cells, especially basophilic normoblasts and polychromatophilic normoblasts, with a decreased level of later-stage erythroblasts such as orthochromic normoblasts [30]. In addition, a changed myeloid to erythroid (M:E) cell ratio of 1:3 in β-thalassemia major as compared to the normal M:E ratio of 1.4:1 [30][31][30,31] has been observed. An erythrokinetic study in β0-thalassemia/HbE patients (six mild and five severe patients) measured RBC terminal half-life and erythrocyte iron utilization by intravenous injection of autologous RBC labeled with 51Cr, and the injection of 59Fe in the form of iron sulfate in donor plasma [3]. The severe β0-thalassemia/HbE patients had a shortened RBC survival in the peripheral blood circulation (9.2 ± 1.9 days) as compared to mild cases (17.0 ± 4.3 days), and both types of patients had shorted RBC survival times as compared to the reference range in normal subjects of 25.7 ± 0.9 days. Interestingly, while there was not a significant difference in erythrocyte iron utilization between severe (35.3 ± 8.9%) and mild cases (35.7 ± 9.0%), these levels were a significant decrease as compared to normal subjects (80%). The low iron utilization seen in β-thalassemia patients is consistent with the occurrence of a massive loss of developing erythroblasts. Though anemia in β-thalassemia is caused by both hemolysis and ineffective erythropoiesis, the erythrokinetic and ferrokinetic analyses indicated that ineffective erythropoiesis plays a much more prominent role than hemolysis. The significant question therefore is what happens to the differentiating erythroblasts in bone marrow of β-thalassemia patients that results in a lack of mature RBCs.
Studies have shown that several factors contribute to ineffective erythropoiesis in β-thalassemia (Figure 3). Apoptosis has been proposed to be the cause of erythroblast death, and accelerated apoptosis of β-thalassemia erythroblasts has been demonstrated both ex vivo in the bone marrow and in vitro in erythroblast culture systems. DNA fragmentation (a hallmark of apoptosis) during the death of β-thalassemia major erythroblasts was shown by the presence of DNA ladders in DNA preparations from CD45− bone marrow erythroblasts [31]. Similarly, bone marrow erythroblasts after culture for 7 and 14 days showed a majority of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)+ basophilic normoblasts and polychromatophilic normoblasts [30]. Hoechst 33342+ erythroblasts have also been detected in cluster of differentiation (CD)45− bone marrow cells of β-thalassemia patients [3]. In addition, increased phosphatidylserine (PS)-exposing basophilic normoblasts and polychromatophilic normoblasts have been reported in β-thalassemic bone marrow erythroblasts from both patients and from a mouse model [3][26][28][3,26,28].
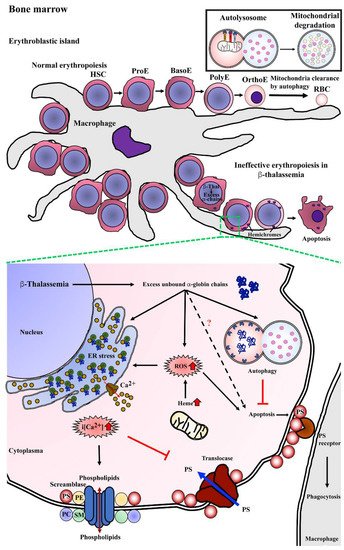
Figure 3. Mechanism of program cell death in bone marrow β-thalassemic erythroblasts. Normal erythropoiesis occurs in bone marrow in the location of an erythroblastic island that contains a macrophage as a feeder cell to product cytokines, which is essential for erythroid differentiation from hematopoietic stem cells (HSCs) to erythroid precursor cells including pronormoblasts (ProE), basophilic normoblasts (BasoE), polychromatophilic normoblasts (PolyE), and orthochromatic normoblasts (OrthoE) and consequently enucleation and mitochondria clearance by autophagy resulting in terminal erythroid differentiation and yielding mature red blood cells (RBCs). In β-thalassemia, excess unbound α-globin chains precipitate into erythroblasts leading to hemichrome accumulation and cellular stress. Autophagy could be a process of cellular adaptation in β-thalassemic erythroblasts to reduce the toxicity from excess unbound α-globin chains by protein degradation and inhibit apoptosis. However, the imbalance of α-globin/non-α-globin and increased heme in erythroblasts cause reactive oxygen species (ROS) generation via the Fenton reaction that consequently induces an ER stress response, resulting in the release of calcium (Ca2+) into cytoplasm. Increased intracellular calcium (i[Ca2+]) effects to activate scramblase but inhibits translocase, leading to the loss of plasma membrane asymmetry, resulting in increased phosphatidylserine (PS) on the outer membrane leaflet. PS-bearing erythroblasts could be cleared by macrophages via PS-PS receptor interaction as the “eat me” signal of phagocytosis.
Defects of β-globin chain synthesis in parallel with normal α-globin synthesis results in the presence of excess unbound α-globin chains. Accumulation and precipitation of excess α-globin chains in thalassemic erythroblast cytoplasm has been demonstrated in bone marrow erythroblasts obtained from β-thalassemia major and β-thalassemia/HbE as determined by electron microscope and laser confocal fluorescence microscopy [31][32][31,32]. The free heme released from the denatured excess α-globin chains could catalyze the formation of reactive oxygen species (ROS), such as hydroxyl radicals via the Fenton reaction, and consequently increase ROS in the thalassemic erythroblast. Bone marrow erythroblasts from the heterozygous βIVS2−654 thalassemic mouse model had a significantly increased α/β-globin ratio and increased ROS levels as compared to wild type mice, and there was a high correlation between the α/β-globin ratio and ROS levels at r = 0.92 [33]. Similar to the in vitro study, β0-thalassemia/HbE erythroblasts obtained from the culture of CD34+ peripheral blood stem cells showed increased ROS levels at day 7, 10, and 14 with basophilic normoblasts, polychromatophilic normoblasts, and orthochromic normoblasts being the majority erythroblast population on each day of culture examined, respectively [34]. The increased ROS can cause the progressive modification or degradation of cellular biomolecules, including DNA, protein, and lipids that could lead to loss of cell function and cell death.
The increased ROS generation is believed to contribute to increased lipid peroxidation, the loss of plasma membrane asymmetry and exposure of phosphatidylserine (PS) on the outer membrane lipid leaflet. In addition, excess unbound α-globin chains in β-thalassemic erythroblasts leads to ER stress. Oxidative stress together with ER stress could influence the flux of Ca2+ out of the ER, and as membrane lipid bilayer asymmetry is maintained by specific lipid transporters, such as amino-phospholipid translocase and phospholipid scramblase, increased Ca2+ can lead to membrane lipid asymmetry through lowered translocase activity and increased phospholipid scramblase activity consequently leading to PS exposure [35]. Increased scramblase activity in β-thalassemia mice erythroid cells has been previously demonstrated, and the majority of thalassemic erythroid cells with PS exposure have activated phospholipid scramblase [36]. Elevated levels of Ca2+ in β-thalassemia/HbE erythroblasts has been reported [37][38][39][37,38,39], and interestingly, reduction of Ca2+ in β-thalassemia/HbE erythroblasts by treatment with EGTA reduced the level of PS exposure to approximately normal control levels [39].
PS exposure on the cell surface is a signal for macrophages to phagocytose that cell, with clearance being needed as a consequence of senescence or damage. Transmission electron microscope analysis of bone marrow macrophages from β-thalassemia major patients showed intracytoplasmic inclusions consisting of phagocytosed erythroblasts and extruded erythroblasts at different stages of degradation [40]. Notably, a two-fold increase in macrophages with phagocytosed β-thalassemic bone marrow erythroblasts as compared to normal bone marrow erythroblasts has been reported [41]. The phagocytosis of β-thalassemic bone marrow erythroblasts was inhibited strongly by annexin V and PS-carrying vesicles, which confirms PS exposure as an “eat me” signal for macrophages [41]. Thus, increased PS exposure and perhaps other features of thalassemic erythroid precursors might lead to their enhanced phagocytic removal as another cause of ineffective erythropoiesis.