Interferon gamma (IFN-γ) is a dimerized soluble cytokine that is the only member of the type II class of interferons. The existence of this interferon, which early in its history was known as immune interferon, was described by E. F. Wheelock as a product of human leukocytes stimulated with phytohemagglutinin, and by others as a product of antigen-stimulated lymphocytes. It was also shown to be produced in human lymphocytes. or tuberculin-sensitized mouse peritoneal lymphocytes challenged with Mantoux test (PPD); the resulting supernatants were shown to inhibit growth of vesicular stomatitis virus. Those reports also contained the basic observation underlying the now widely employed IFN-γ release assay used to test for tuberculosis. In humans, the IFN-γ protein is encoded by the IFNG gene.
- vesicular stomatitis
- phytohemagglutinin
- peritoneal
- interferon
1. Function
IFN-γ, or type II interferon, is a cytokine that is critical for innate and adaptive immunity against viral, some bacterial and protozoan infections. IFN-γ is an important activator of macrophages and inducer of major histocompatibility complex class II molecule expression. Aberrant IFN-γ expression is associated with a number of autoinflammatory and autoimmune diseases. The importance of IFN-γ in the immune system stems in part from its ability to inhibit viral replication directly, and most importantly from its immunostimulatory and immunomodulatory effects. IFN-γ is produced predominantly by natural killer cells (NK) and natural killer T cells (NKT) as part of the innate immune response, and by CD4 Th1 and CD8 cytotoxic T lymphocyte (CTL) effector T cells once antigen-specific immunity develops[1][2] as part of the adaptive immune response. IFN-γ is also produced by non-cytotoxic innate lymphoid cells (ILC), a family of immune cells first discovered in the early 2010s.[3]
2. Structure
The IFN-γ monomer consists of a core of six α-helices and an extended unfolded sequence in the C-terminal region.[4][5] This is shown in the structural models below. The α-helices in the core of the structure are numbered 1 to 6.
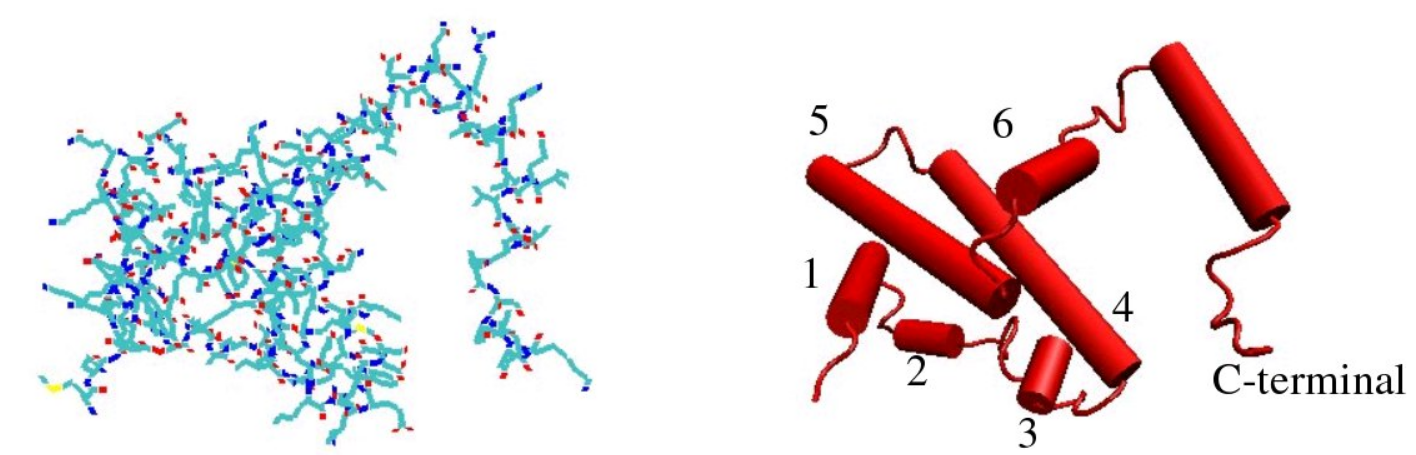
The biologically active dimer is formed by anti-parallel inter-locking of the two monomers as shown below. In the cartoon model, one monomer is shown in red, the other in blue.
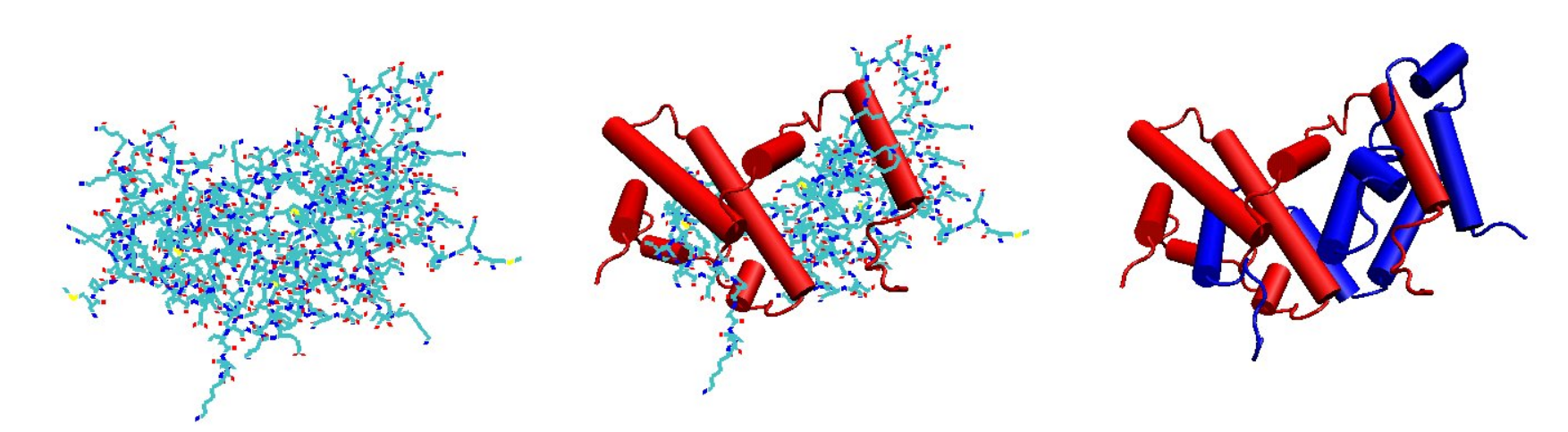
3. Receptor Binding
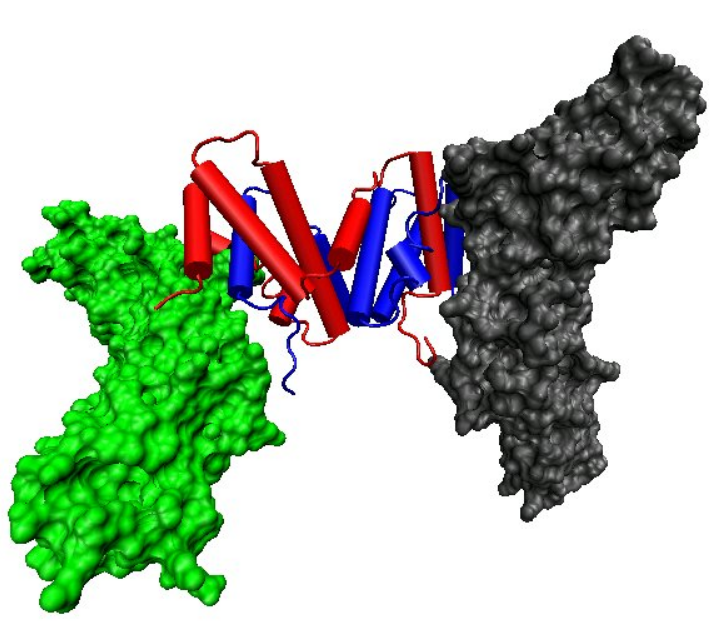
- Cellular responses to IFN-γ are activated through its interaction with a heterodimeric receptor consisting of Interferon gamma receptor 1 (IFNGR1) and Interferon gamma receptor 2 (IFNGR2). IFN-γ binding to the receptor activates the JAK-STAT pathway. Activation of the JAK-STAT pathway induces upregulation of interferon-stimulated genes (ISGs), including MHC II.[6] IFN-γ also binds to the glycosaminoglycan heparan sulfate (HS) at the cell surface. However, in contrast to many other heparan sulfate binding proteins, where binding promotes biological activity, the binding of IFN-γ to HS inhibits its biological activity.[7]
The structural models shown in figures 1-3 for IFN-γ[5] are all shortened at their C-termini by 17 amino acids. Full length IFN-γ is 143 amino acids long, the models are 126 amino acids long. Affinity for heparan sulfate resides solely within the deleted sequence of 17 amino acids.[8] Within this sequence of 17 amino acids lie two clusters of basic amino acids termed D1 and D2, respectively. Heparan sulfate interacts with both of these clusters.[9] In the absence of heparan sulfate the presence of the D1 sequence increases the rate at which IFN-γ-receptor complexes form.[7] Interactions between the D1 cluster of amino acids and the receptor may be the first step in complex formation. By binding to D1 HS may compete with the receptor and prevent active receptor complexes from forming.
The biological significance of heparan sulfates interaction with IFN-γ is unclear; however, binding of the D1 cluster to HS may protect it from proteolytic cleavage.[9]
4. Biological Activity
IFN-γ is secreted by T helper cells (specifically, Th1 cells), cytotoxic T cells (TC cells), macrophages, mucosal epithelial cells and NK cells. IFN-γ is both an important autocrine signal for professional APCs in early innate immune response, and an important paracrine signal in adaptive immune response. The expression of IFN-γ is induced by the cytokines IL-12, IL-15, IL-18, and type I IFN.[10] IFN-γ is the only Type II interferon and it is serologically distinct from Type I interferons; it is acid-labile, while the type I variants are acid-stable.
IFN-γ has antiviral, immunoregulatory, and anti-tumor properties.[11] It alters transcription in up to 30 genes producing a variety of physiological and cellular responses. Among the effects are:
- Promotes NK cell activity[12]
- Increases antigen presentation and lysosome activity of macrophages.
- Activates inducible nitric oxide synthase (iNOS)
- Induces the production of IgG2a and IgG3 from activated plasma B cells
- Causes normal cells to increase expression of class I MHC molecules as well as class II MHC on antigen-presenting cells—to be specific, through induction of antigen processing genes, including subunits of the immunoproteasome (MECL1, LMP2, LMP7), as well as TAP and ERAAP in addition possibly to the direct upregulation of MHC heavy chains and B2-microglobulin itself
- Promotes adhesion and binding required for leukocyte migration
- Induces the expression of intrinsic defense factors—for example, with respect to retroviruses, relevant genes include TRIM5alpha, APOBEC, and Tetherin, representing directly antiviral effects
- Primes alveolar macrophages against secondary bacterial infections.[13][14]
IFN-γ is the primary cytokine that defines Th1 cells: Th1 cells secrete IFN-γ, which in turn causes more undifferentiated CD4+ cells (Th0 cells) to differentiate into Th1 cells ,[15] representing a positive feedback loop—while suppressing Th2 cell differentiation. (Equivalent defining cytokines for other cells include IL-4 for Th2 cells and IL-17 for Th17 cells.)
NK cells and CD8+ cytotoxic T cells also produce IFN-γ. IFN-γ suppresses osteoclast formation by rapidly degrading the RANK adaptor protein TRAF6 in the RANK-RANKL signaling pathway, which otherwise stimulates the production of NF-κB.
4.1. Activity in Granuloma Formation
A granuloma is the body's way of dealing with a substance it cannot remove or sterilize. Infectious causes of granulomas (infections are typically the most common cause of granulomas) include tuberculosis, leprosy, histoplasmosis, cryptococcosis, coccidioidomycosis, blastomycosis, and toxoplasmosis. Examples of non-infectious granulomatous diseases are sarcoidosis, Crohn's disease, berylliosis, giant-cell arteritis, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, pulmonary rheumatoid nodules, and aspiration of food and other particulate material into the lung.[16] The infectious pathophysiology of granulomas is discussed primarily here.
The key association between IFN-γ and granulomas is that IFN-γ activates macrophages so that they become more powerful in killing intracellular organisms.[17] Activation of macrophages by IFN-γ from Th1 helper cells in mycobacterial infections allows the macrophages to overcome the inhibition of phagolysosome maturation caused by mycobacteria (to stay alive inside macrophages).[18][19] The first steps in IFN-γ-induced granuloma formation are activation of Th1 helper cells by macrophages releasing IL-1 and IL-12 in the presence of intracellular pathogens, and presentation of antigens from those pathogens. Next the Th1 helper cells aggregate around the macrophages and release IFN-γ, which activates the macrophages. Further activation of macrophages causes a cycle of further killing of intracellular bacteria, and further presentation of antigens to Th1 helper cells with further release of IFN-γ. Finally, macrophages surround the Th1 helper cells and become fibroblast-like cells walling off the infection.
4.2. Activity during Pregnancy
Uterine Natural Killer cells (NK) secrete high levels of chemoattractants, such as IFN-γ in mice. IFN-γ dilates and thins the walls of maternal spiral arteries to enhance blood flow to the implantation site. This remodeling aids in the development of the placenta as it invades the uterus in its quest for nutrients. IFN-γ knockout mice fail to initiate normal pregnancy-induced modification of decidual arteries. These models display abnormally low amounts of cells or necrosis of decidua.[20]
In humans, elevated levels of IFN-γ have been associated with increased risk of miscarriage. Correlation studies have observed high IFN-γ levels in women with a history of spontaneous miscarriage, when compared to women with no history of spontaneous miscarriage.[21] Additionally, low-IFN-γ levels are associated with women who successfully carry to term. It is possible that IFN-γ is cytotoxic to trophoblasts, which leads to miscarriage.[22] However, causal research on the relationship between IFN-γ and miscarriage has not been performed due to ethical constraints.
5. Production
Recombinant human IFN-γ, as an expensive biopharmaceutical, has been expressed in different expression systems including prokaryotic, protozoan, fungal (yeasts), plant, insect and mammalian cells. Human IFN-γ is commonly expressed in Escherichia coli, marketed as ACTIMMUNE®, however, the resulting product of the prokaryotic expression system is not glycosylated with a short half-life in the bloodstream after injection; the purification process from bacterial expression system is also very costly. Other expression systems like Pichia pastoris did not show satisfactory results in terms of yields.[23][24]
6. Therapeutic Use
Interferon-γ 1b is approved by the U.S. Food and Drug Administration to treat chronic granulomatous disease[25] (CGD) and osteopetrosis.[26] The mechanism by which IFN-γ benefits CGD is via enhancing the efficacy of neutrophils against catalase-positive bacteria by correcting patients' oxidative metabolism.[27]
It was not approved to treat idiopathic pulmonary fibrosis (IPF). In 2002, the manufacturer InterMune issued a press release saying that phase III data demonstrated survival benefit in IPF and reduced mortality by 70% in patients with mild to moderate disease. The U.S. Department of Justice charged that the release contained false and misleading statements. InterMune's chief executive, Scott Harkonen, was accused of manipulating the trial data, was convicted in 2009 of wire fraud, and was sentenced to fines and community service. Harkonen appealed his conviction to the U.S. Court of Appeals for the Ninth Circuit, and lost.[28] Harkonen was granted a full pardon on January 20, 2021.[29]
Preliminary research on the role of IFN-γ in treating Friedreich's ataxia (FA) conducted by Children’s Hospital of Philadelphia has found no beneficial effects in short-term (< 6-months) treatment.[30][31][32] However, researchers in Turkey have discovered significant improvements in patients' gait and stance after 6 months of treatment.[33]
Although not officially approved, Interferon-γ has also been shown to be effective in treating patients with moderate to severe atopic dermatitis.[34][35][36] Specifically, recombinant IFN-γ therapy has shown promise in patients with lowered IFN-γ expression, such as those with predisposition to herpes simplex virus, and pediatric patients.[37]
7. Potential Use in Immunotherapy
IFN-γ increases an anti-proliferative state in cancer cells, while upregulating MHC I and MHC II expression, which increases immunorecognition and removal of pathogenic cells.[38] IFN-γ also reduces metastasis in tumors by upregulating fibronectin, which negatively impacts tumor architecture.[39]
IFN-γ is not approved yet for the treatment in any cancer immunotherapy. However, improved survival was observed when IFN-γ was administrated to patients with bladder carcinoma and melanoma cancers. The most promising result was achieved in patients with stage 2 and 3 of ovarian carcinoma. On the contrary, it was stressed: "Interferon-γ secreted by CD8-positive lymphocytes upregulates PD-L1 on ovarian cancer cells and promotes tumour growth."[40] The in vitro study of IFN-γ in cancer cells is more extensive and results indicate anti-proliferative activity of IFN-γ leading to the growth inhibition or cell death, generally induced by apoptosis but sometimes by autophagy.[23] In addition, it has been reported that mammalian glycosylation of recombinant human IFN-γ, expressed in HEK293, improves its therapeutic efficacy compared to the unglycosylated form that is expressed in E. coli.[41]
8. Interactions
Interferon-γ has been shown to interact with Interferon gamma receptor 1 and Interferon gamma receptor 2.[42][43]
8.1. Diseases
Interferon-γ has been shown to be a crucial player in the immune response against some intracellular pathogens, including that of Chagas disease.[44] It has also been identified as having a role in seborrheic dermatitis.[45]
IFN-γ has a significant anti-viral effect in herpes simplex virus I (HSV) infection. IFN-γ compromises the microtubules that HSV relies upon for transport into an infected cell's nucleus, inhibiting the ability of HSV to replicate.[46][47] Studies in mice on acyclovir resistant herpes have shown that IFN-γ treatment can significantly reduce herpes viral load. The mechanism by which IFN-γ inhibits herpes reproduction is independent of T-cells, which means that IFN-γ may be an effective treatment in individuals with low T-cells.[48][49][50]
Chlamydia infection is impacted by IFN-γ in host cells. In human epithelial cells, IFN-γ upregulates expression of indoleamine 2,3-dioxygenase, which in turn depletes tryptophan in hosts and impedes chlamydia's reproduction.[51][52] Additionally, in rodent epithelial cells, IFN-γ upregulates a GTPase that inhibits chlamydial proliferation.[53] In both the human and rodent systems, chlamydia has evolved mechanisms to circumvent the negative effects of host cell behavior.[54]
9. Regulation
There is evidence that interferon-gamma expression is regulated by a pseudoknotted element in its 5' UTR.[55] There is also evidence that interferon-gamma is regulated either directly or indirectly by the microRNAs: miR-29.[56] Furthermore, there is evidence that interferon-gamma expression is regulated via GAPDH in T-cells. This interaction takes place in the 3'UTR, where binding of GAPDH prevents the translation of the mRNA sequence.[57]
References
- "Entrez Gene: INFG". https://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=3458.
- "Regulation of Interferon‐γ During Innate and Adaptive Immune Responses". Regulation of interferon-gamma during innate and adaptive immune responses. 96. 2007. 41–101. doi:10.1016/S0065-2776(07)96002-2. ISBN 978-0-12-373709-0. https://dx.doi.org/10.1016%2FS0065-2776%2807%2996002-2
- "The biology of innate lymphoid cells". Nature 517 (7534): 293–301. January 2015. doi:10.1038/nature14189. PMID 25592534. Bibcode: 2015Natur.517..293A. https://dx.doi.org/10.1038%2Fnature14189
- "Three-dimensional structure of recombinant human interferon-gamma". Science 252 (5006): 698–702. May 1991. doi:10.1126/science.1902591. PMID 1902591. Bibcode: 1991Sci...252..698E. https://dx.doi.org/10.1126%2Fscience.1902591
- PDB: 1FG9; "Observation of an unexpected third receptor molecule in the crystal structure of human interferon-gamma receptor complex". Structure 8 (9): 927–936. September 2000. doi:10.1016/S0969-2126(00)00184-2. PMID 10986460. https://www.rcsb.org/structure/1FG9
- "Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases". Immunity 31 (4): 539–550. October 2009. doi:10.1016/j.immuni.2009.09.002. PMID 19833085. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2774226
- "The heparan sulfate binding sequence of interferon-gamma increased the on rate of the interferon-gamma-interferon-gamma receptor complex formation". The Journal of Biological Chemistry 273 (18): 10919–10925. May 1998. doi:10.1074/jbc.273.18.10919. PMID 9556569. https://dx.doi.org/10.1074%2Fjbc.273.18.10919
- "NMR characterization of the interaction between the C-terminal domain of interferon-gamma and heparin-derived oligosaccharides". The Biochemical Journal 384 (Pt 1): 93–99. November 2004. doi:10.1042/BJ20040757. PMID 15270718. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1134092
- "Interferon-gamma binds to heparan sulfate by a cluster of amino acids located in the C-terminal part of the molecule". FEBS Letters 280 (1): 152–154. March 1991. doi:10.1016/0014-5793(91)80225-R. PMID 1901275. https://dx.doi.org/10.1016%2F0014-5793%2891%2980225-R
- "Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion". Frontiers in Immunology 9: 847. 2018. doi:10.3389/fimmu.2018.00847. PMID 29780381. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5945880
- "Interferon-gamma: an overview of signals, mechanisms and functions". Journal of Leukocyte Biology 75 (2): 163–189. February 2004. doi:10.1189/jlb.0603252. PMID 14525967. https://dx.doi.org/10.1189%2Fjlb.0603252
- "The role of cytokines in the regulation of NK cells in the tumor environment". Cytokine 117: 30–40. May 2019. doi:10.1016/j.cyto.2019.02.001. PMID 30784898. https://dx.doi.org/10.1016%2Fj.cyto.2019.02.001
- "Tissue-Specific Macrophage Responses to Remote Injury Impact the Outcome of Subsequent Local Immune Challenge". Immunity 51 (5): 899–914.e7. November 2019. doi:10.1016/j.immuni.2019.10.010. PMID 31732166. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6892583
- "Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity". Cell 175 (6): 1634–1650.e17. November 2018. doi:10.1016/j.cell.2018.09.042. PMID 30433869. https://dx.doi.org/10.1016%2Fj.cell.2018.09.042
- "CD4⁺T cells: differentiation and functions". Clinical & Developmental Immunology 2012: 925135. 2012. doi:10.1155/2012/925135. PMID 22474485. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3312336
- "Causes of pulmonary granulomas: a retrospective study of 500 cases from seven countries". Journal of Clinical Pathology 65 (1): 51–57. January 2012. doi:10.1136/jclinpath-2011-200336. PMID 22011444. https://dx.doi.org/10.1136%2Fjclinpath-2011-200336
- "IFN-γ primes macrophage activation by increasing phosphatase and tensin homolog via downregulation of miR-3473b". Journal of Immunology 193 (6): 3036–3044. September 2014. doi:10.4049/jimmunol.1302379. PMID 25092892. https://dx.doi.org/10.4049%2Fjimmunol.1302379
- "Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis". PLOS ONE 6 (5): e19105. May 2011. doi:10.1371/journal.pone.0019105. PMID 21559306. Bibcode: 2011PLoSO...619105H. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3085516
- "Th1-Th2 polarisation and autophagy in the control of intracellular mycobacteria by macrophages". Veterinary Immunology and Immunopathology 128 (1–3): 37–43. March 2009. doi:10.1016/j.vetimm.2008.10.293. PMID 19026454. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2789833
- "Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy". The Journal of Experimental Medicine 192 (2): 259–270. July 2000. doi:10.1084/jem.192.2.259. PMID 10899912. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2193246
- "The role of interferons in early pregnancy". Gynecological Endocrinology 30 (1): 1–6. January 2014. doi:10.3109/09513590.2012.743011. PMID 24188446. https://dx.doi.org/10.3109%2F09513590.2012.743011
- "Effects of products of activated leukocytes (lymphokines and monokines) on the growth of malignant trophoblast cells in vitro". American Journal of Obstetrics and Gynecology 158 (1): 199–203. January 1988. doi:10.1016/0002-9378(88)90810-1. PMID 2447775. https://dx.doi.org/10.1016%2F0002-9378%2888%2990810-1
- "Review of the recombinant human interferon gamma as an immunotherapeutic: Impacts of production platforms and glycosylation". Journal of Biotechnology 240: 48–60. December 2016. doi:10.1016/j.jbiotec.2016.10.022. PMID 27794496. https://dx.doi.org/10.1016%2Fj.jbiotec.2016.10.022
- "Is Pichia pastoris a realistic platform for industrial production of recombinant human interferon gamma?". Biologicals 45: 52–60. January 2017. doi:10.1016/j.biologicals.2016.09.015. PMID 27810255. https://zenodo.org/record/1312349.
- "Interferon gamma-1b. A review of its pharmacology and therapeutic potential in chronic granulomatous disease". Drugs 43 (1): 111–122. January 1992. doi:10.2165/00003495-199243010-00008. PMID 1372855. https://dx.doi.org/10.2165%2F00003495-199243010-00008
- "Recombinant human interferon gamma therapy for osteopetrosis". The Journal of Pediatrics 121 (1): 119–124. July 1992. doi:10.1016/s0022-3476(05)82557-0. PMID 1320672. https://dx.doi.org/10.1016%2Fs0022-3476%2805%2982557-0
- "The use of interferon-gamma therapy in chronic granulomatous disease". Recent Patents on Anti-Infective Drug Discovery 3 (3): 225–230. November 2008. doi:10.2174/157489108786242378. PMID 18991804. https://dx.doi.org/10.2174%2F157489108786242378
- "Drug Marketing. The line between scientific uncertainty and promotion of snake oil". BMJ 347: f5687. September 2013. doi:10.1136/bmj.f5687. PMID 24055923. https://dx.doi.org/10.1136%2Fbmj.f5687
- "Statement from the Press Secretary Regarding Executive Grants of Clemency". whitehouse.gov. January 20, 2021. https://trumpwhitehouse.archives.gov/briefings-statements/statement-press-secretary-regarding-executive-grants-clemency-012021/.
- "IFN-γ for Friedreich ataxia: present evidence". Neurodegenerative Disease Management 5 (6): 497–504. December 2015. doi:10.2217/nmt.15.52. PMID 26634868. https://dx.doi.org/10.2217%2Fnmt.15.52
- "Open-label pilot study of interferon gamma-1b in Friedreich ataxia". Acta Neurologica Scandinavica 132 (1): 7–15. July 2015. doi:10.1111/ane.12337. PMID 25335475. https://dx.doi.org/10.1111%2Fane.12337
- "Randomized, double-blind, placebo-controlled study of interferon-γ 1b in Friedreich Ataxia". Annals of Clinical and Translational Neurology 6 (3): 546–553. March 2019. doi:10.1002/acn3.731. PMID 30911578. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=6414489
- "Efficacy and Tolerability of Interferon Gamma in Treatment of Friedreich's Ataxia: Retrospective Study". Noro Psikiyatri Arsivi 57 (4): 270–273. December 2020. doi:10.29399/npa.25047. PMID 33354116. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=7735154
- "Atopic dermatitis: systemic immunosuppressive therapy". Seminars in Cutaneous Medicine and Surgery 27 (2): 151–155. June 2008. doi:10.1016/j.sder.2008.04.004. PMID 18620137. https://dx.doi.org/10.1016%2Fj.sder.2008.04.004
- "Long-term therapy with recombinant interferon-gamma (rIFN-gamma) for atopic dermatitis". Annals of Allergy, Asthma & Immunology 80 (3): 263–268. March 1998. doi:10.1016/S1081-1206(10)62968-7. PMID 9532976. https://dx.doi.org/10.1016%2FS1081-1206%2810%2962968-7
- "Recombinant interferon gamma therapy for atopic dermatitis". Journal of the American Academy of Dermatology 28 (2 Pt 1): 189–197. February 1993. doi:10.1016/0190-9622(93)70026-p. PMID 8432915. https://dx.doi.org/10.1016%2F0190-9622%2893%2970026-p
- "Recent considerations in the use of recombinant interferon gamma for biological therapy of atopic dermatitis". Expert Opinion on Biological Therapy 16 (4): 507–514. 2016. doi:10.1517/14712598.2016.1135898. PMID 26694988. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4985031
- "Interferon-gamma (IFN-γ): Exploring its implications in infectious diseases". Biomolecular Concepts 9 (1): 64–79. May 2018. doi:10.1515/bmc-2018-0007. PMID 29856726. https://dx.doi.org/10.1515%2Fbmc-2018-0007
- "Roles of IFN-γ in tumor progression and regression: a review". Biomarker Research 8 (1): 49. 2020-09-29. doi:10.1186/s40364-020-00228-x. PMID 33005420. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=7526126
- "IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer". British Journal of Cancer 112 (9): 1501–1509. April 2015. doi:10.1038/bjc.2015.101. PMID 25867264. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4453666
- "Improved therapeutic efficacy of mammalian expressed-recombinant interferon gamma against ovarian cancer cells". Experimental Cell Research 359 (1): 20–29. October 2017. doi:10.1016/j.yexcr.2017.08.014. PMID 28803068. https://dx.doi.org/10.1016%2Fj.yexcr.2017.08.014
- "Observation of an unexpected third receptor molecule in the crystal structure of human interferon-gamma receptor complex". Structure 8 (9): 927–936. September 2000. doi:10.1016/S0969-2126(00)00184-2. PMID 10986460. https://dx.doi.org/10.1016%2FS0969-2126%2800%2900184-2
- "Interaction between the components of the interferon gamma receptor complex". The Journal of Biological Chemistry 270 (36): 20915–20921. September 1995. doi:10.1074/jbc.270.36.20915. PMID 7673114. https://dx.doi.org/10.1074%2Fjbc.270.36.20915
- "IL18 Gene Variants Influence the Susceptibility to Chagas Disease". PLOS Neglected Tropical Diseases 10 (3): e0004583. March 2016. doi:10.1371/journal.pntd.0004583. PMID 27027876. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4814063
- "Investigations of seborrheic dermatitis. Part I. The role of selected cytokines in the pathogenesis of seborrheic dermatitis". Postepy Higieny I Medycyny Doswiadczalnej 66: 843–847. November 2012. doi:10.5604/17322693.1019642. PMID 23175340. https://dx.doi.org/10.5604%2F17322693.1019642
- "Complexity of Interferon-γ Interactions with HSV-1". Frontiers in Immunology 5: 15. 2014-02-06. doi:10.3389/fimmu.2014.00015. PMID 24567732. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3915238
- "Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus". The Journal of Cell Biology 136 (5): 1007–1021. March 1997. doi:10.1083/jcb.136.5.1007. PMID 9060466. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2132479
- "Beta interferon plus gamma interferon efficiently reduces acyclovir-resistant herpes simplex virus infection in mice in a T-cell-independent manner". The Journal of General Virology 91 (Pt 3): 591–598. March 2010. doi:10.1099/vir.0.016964-0. PMID 19906941. https://dx.doi.org/10.1099%2Fvir.0.016964-0
- "Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1". Journal of Virology 76 (22): 11541–11550. November 2002. doi:10.1128/JVI.76.22.11541-11550.2002. PMID 12388715. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=136787
- "Immune control of herpes simplex virus during latency". Current Opinion in Immunology 16 (4): 463–469. August 2004. doi:10.1016/j.coi.2004.05.003. PMID 15245740. https://dx.doi.org/10.1016%2Fj.coi.2004.05.003
- "The role of IFN-gamma in the outcome of chlamydial infection". Current Opinion in Immunology 14 (4): 444–451. August 2002. doi:10.1016/s0952-7915(02)00361-8. PMID 12088678. https://dx.doi.org/10.1016%2Fs0952-7915%2802%2900361-8
- "Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism". FASEB Journal 5 (11): 2516–2522. August 1991. doi:10.1096/fasebj.5.11.1907934. PMID 1907934. https://dx.doi.org/10.1096%2Ffasebj.5.11.1907934
- "The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice". Proceedings of the National Academy of Sciences of the United States of America 103 (38): 14092–14097. September 2006. doi:10.1073/pnas.0603338103. PMID 16959883. Bibcode: 2006PNAS..10314092B. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1599917
- "Chlamydial IFN-gamma immune evasion is linked to host infection tropism". Proceedings of the National Academy of Sciences of the United States of America 102 (30): 10658–10663. July 2005. doi:10.1073/pnas.0504198102. PMID 16020528. Bibcode: 2005PNAS..10210658N. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1180788
- "Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR". Cell 108 (2): 221–232. January 2002. doi:10.1016/S0092-8674(02)00616-5. PMID 11832212. https://dx.doi.org/10.1016%2FS0092-8674%2802%2900616-5
- "MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components". Molecular Immunology 45 (7): 1995–2006. April 2008. doi:10.1016/j.molimm.2007.10.035. PMID 18061676. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2678893
- "Posttranscriptional control of T cell effector function by aerobic glycolysis". Cell 153 (6): 1239–1251. June 2013. doi:10.1016/j.cell.2013.05.016. PMID 23746840. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3804311