Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Beatrix Zheng and Version 3 by Beatrix Zheng.
Cyclic nucleotides (cAMP, cGMP) play a major role in normal and pathologic signaling. Beyond receptors, cyclic nucleotide phosphodiesterases; (PDEs) rapidly convert the cyclic nucleotide in its respective 5′-nucleotide to control intracellular cAMP and/or cGMP levels to maintain a normal physiological state. However, in many pathologies, dysregulations of various PDEs (PDE1-PDE11) contribute mainly to organs and tissue failures related to uncontrolled phosphorylation cascade. Among these, PDE4 represents the greatest family, since it is constituted by 4 genes with multiple variants differently distributed at tissue, cellular and subcellular levels, allowing different fine-tuned regulations.
- cyclic nucleotide phosphodiesterase (PDE)
- cAMP
- cGMP
- diseases
1. PDE4 in Cardiovascular Diseases
PDE4 participates in the control of cardiovascular function at the level of vessels and heart.
1.1. PDE4, and Vessel
PDE4 has been characterized in human, rat, and bovine aortas [1]. It is clearly established that in vascular smooth muscle, PDE4 inhibitors control endothelium-dependent relaxation, opposite to PDE3 inhibitors or nitric oxide (NO) that per se relax the vessels [2][3][4]. This is related to the different kinetic properties of PDE3 and PDE4 (PDE3 Km < PDE4 Km, and PDE3 Vmax/Km > PDE4 Vmax/Km) as illustrated in Figure 1. In the presence of endothelium, at low intracellular cAMP concentrations, firstly PDE3 hydrolyses cAMP; therefore, its specific inhibition, or the endothelial NO production, induces an increase in the cAMP level thus allowing PDE4 hydrolytic function. In that condition, the inhibition of PDE4 would be effective by increasing the cAMP level and would therefore relax the whole vessel. Altogether, at the level of vascular smooth muscle, the researchers demonstrated the existence of cAMP/cGMP cross-talk mediated by PDE3 and PDE4 [4][5][6].
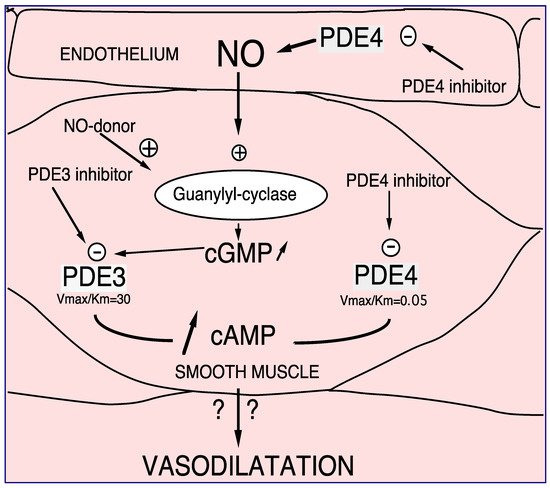
Figure 1. PDE3 and PDE4 cross-talk in vasodilation. NO or NO donor, by increasing soluble guanylyl cyclase, increases intracellular cGMP level. Therefore, this increase in cGMP induces preferentially PDE3 inhibition and consequently, an increase in cAMP in smooth muscle cells. In that way PDE4 might be able to hydrolyze cAMP in that way, PDE4 inhibitors inhibit PDE4 activity. In the presence of endothelial cells this effect is potentiated. Altogether, increases in cAMP altogether induces vasodilatation which might be potentiated in the presence of endothelium [2][3][4][5].
Moreover, it should be noted that PDE4 is mainly present in human endothelial cells [7][8][9][10]. Surprisingly, in the presence of L-arginine, the PDE4 inhibitor, rolipram, is able to increase cGMP levels by upregulating the L-arginine/NO/cGMP pathway, revealing a cross-talk between PDE4 and cGMP regulation [11] (Figure 2).
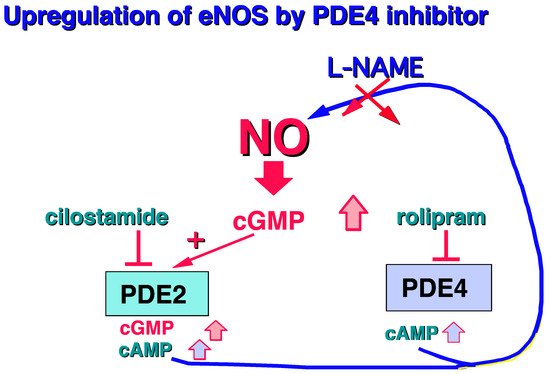
Figure 2. Increase in cGMP in human endothelial cells related to PDE4 inhibition [11].
In particular, PDE4 plays a role in the regulation of endothelial permeability [12]. Notably the PDE4D, by tethering the exchange protein activated by cAMP (EPAC)1 in a vascular endothelial cadherin-based signaling complex (VE-Cad), controls the cAMP-mediated vascular permeability [13]. It should be noted that heart failure in rat induces increases in the PDE4B protein and mRNA expressions in the aorta which could contribute to high blood pressure [14]. Lastly, it was shown that PDE4B, and not PDE4D, was upregulated in inflammatory cells from both experimental and human abdominal aortic aneurism, limiting the progressive increase in the aortic diameter without affecting the blood pressure. Rolipram has strongly mitigated the increase in vascular oxidative stress (superoxide anion) induced by angiotensin II [15]. Clinically, it is important to note that PDE4 inhibition reduces life-sustaining extracorporeal vascular permeability and improves microcirculation in an extracorporeal resuscitation rodent model [16].
1.2. PDE4, and Heart
The cAMP in cardiac muscle is the second intracellular messenger mediating the positive inotrope effects of β-agonists [17]. Thus, PDE3 inhibitors, such as milrinone, SK, and F 94120, were described early in the early 1980s as “new cardiotonic drugs” [18][19]. Consequently, it was clearly established that the positive inotrope effect induced by PDE3 inhibitors was mediated by cAMP [20]. However, due to some mortality in chronic treatment related to tachyarrhythmia and tachycardia, milrinone prescription in humans is now only performed for a short period, beneficially inducing reduced inflammatory and apoptotic signaling [21]. At the same time, a rolipram-sensitive cAMP-PDE has been characterized in the heart. This was carried out by anion exchange chromatography performed on cardiac tissues from the rat heart ventricle [22], as well as from canine left ventricles [23] and from frog atrial fibers [24]. Furthermore, Komas et al., 1989 [19] isolated by chromatography the rolipram-inhibited cAMP-PDE (PDE4) from dog ventricular and sino-atrial node tissues, pointing out different inhibitory effects of specific inhibitors between the ventricle and sino-atrial node phosphodiesterases (PDEs). Thus, the characterization of PDE4 in cardiac tissues raised the question of its implication in cardiac contraction.
Studies performed on guinea pig cardiac ventricle showed that PDE4 might be implicated differently than PDE3 in the regulation of cardiac contraction, since in opposite to PDE3 inhibitor, PDE4 inhibitor did not per se increase cardiac inotropy, although PDE3 and PDE4 are both cytosolic and membrane bound [25][26]. However, interestingly, PDE4 inhibitors potentiate the effect of PDE3 inhibition, showing that in the presence of cyclic AMP-dependent positive inotropic agents, PDE4 inhibitors exert a positive inotropic effect which probably does not involve enhanced catecholamine release from sympathetic nerve endings [26]. This was confirmed more recently by Eschenhagen, 2013 [27] indicating that rolipram does not affect the positive inotropic effects from β1- or β2-adrenoceptor stimulation. This might also suggest that PDE4 acts in another cAMP compartment.
Thus, the researchers firstly showed in canine and human, purified cardiac microsomal fractions, that PDE3 is associated with the microsomal membranes enriched in vesicles derived from T-tubule and junctional SR membranes; meanwhile, PDE4 is mostly associated with the enriched fraction of sarcolemma membranes, revealing different subcellular compartmentations for PDE3 and PDE4 [28]. To go further, the researchers have canonically demonstrated that PDE4B and PDE4D are associated with the envelope of the nucleus isolated from human cardiac cells [29]. This has only been confirmed very recently by a pharmacological approach, demonstrating that PDE4 insulates a mAKAPβ-targeted PKA pool at the nuclear envelope [30]. In that way, Marco Conti and his colleagues showed that PDE4D isoforms are anchored by myomegalin colocalizing components of the cAMP-dependent pathway to the Golgi/centrosomal region of the cardiac cell [31]. Due to the different physiological and localized contributions of PDE4 in cardiac contraction, its physiopathological role in cardiac pathology could therefore be questioned.
2. PDE4 in Obesity
Obesity is considered the fifth highest cause of death worldwide by the World Health Organization and is associated with pathological conditions and diseases associated with obesity, including hyperlipidemia, heart diseases such as coronary artery disease (CAD) and myocardial infarction, stroke, type 2 diabetes (T2D), hypertension, cancers, low-grade and chronic inflammation, fatty liver disease, osteoarthritis, respiratory problems, and neurodegenerative diseases [32].
Obesity is defined by the body mass index (BMI). It is calculated as weight in kilograms divided by height in meters squared, rounded to one decimal place. Obesity in adults (>20 years old) was defined as a BMI greater than or equal to 30. The prevalence of obesity was 39.8% and affected about 93.3 million US adults in 2015~2016 [33].
Therefore, it was pointed out that caloric restriction might extend lifespan and that the metabolic effect of resveratrol, a mimetic of caloric restriction, might be mediated by inhibiting cAMP phosphodiesterases [34]. In that way, alterations in PDE activities were firstly reported in omental and subcutaneous adipose tissues in human obesity, i.e., in omental and subcutaneous adipose tissues, it was a significant negative correlation between PDE4 and BMI [35].
Thus, the involvement of PDE4 in obesity has been demonstrated since, under submaximal β-adrenoceptor stimulation of brown adipocytes, the PDE4 inhibitor alone may increase lipolysis [36]. Therefore, it was reported that the PDE4 inhibitor, roflumilast, might be superior to metformin in weight loss in obese women with polycystic ovarian fibrosis [37]. In this way, PDE4B-KO mice had reduced adiposity and adipose-induced inflammation induced by a high-fat diet, highlighting the involvement of PDE4B in adiposity [38].
3. PDE4 in Diabetes
The global prevalence of diabetes among 20- to 79-year-olds is estimated to be about 8.8% in 2015, or 415 million, and approximately 10.4% in 2040, or 642 million [39]. Diabetes is related to alterations in glucose homeostasis governed by insulin. The failure to maintain glucose homeostasis underlies both type 1 diabetes (T1D) and type 2 diabetes (T2D). T1D is an autoimmune disease that originates when β-cells that produce insulin are destroyed. T2D is characterized by insulin resistance and the progressive loss of β-cell function. Although these two forms of diabetes are fundamentally very different, β-cell failure and death play a key role in the pathogenesis of both diseases, leading to hyperglycemia resulting from a reduced capacity to produce insulin [40]. The diagnosis of diabetes and its severity may be determined by measuring blood glycated proteins such as hemoglobin A1c (HbA1c) and glycated albumin (GA) [41]. In the 2000s, it was proposed that inhibiting PDE4 during diabetes may be beneficial against hyperglycemia, oxidative stress, and the production of TNF-α and NFκB [42]. In this way, it has previously been reported that insulin-secreting cells and Langerhans islets contain PDE4 [43]. PDE4C is the main PDE4 subtype expressed at the mRNA level in isolated rat islets. Interestingly, silencing the PDE4C as well as the specific PDE4 inhibitors, roflumilast and compound L-826,141, significantly increases glucose-dependent insulin secretion [44]. PDE4 seemed active only after stimulation with glucose, suggesting some interaction between glucose and PDE4 [45]. Following this, a study in mice showed that roflumilast improves glucose tolerance and insulin sensitivity [45].
4. PDE4 and Ulcerative Colitis and Crohn’Disease
Crohn’s disease is a chronic inflammatory condition of the gastrointestinal tract. It can affect any part of the gastrointestinal (GI) tract, but ulcerative colitis affects only the colon. Additionally, while Crohn’s disease can affect all layers of the bowel wall, ulcerative colitis (UC) only affects the lining of the colon. Both have been together classified as inflammatory bowel diseases (IBD). In the United States, it is currently estimated that about 1.5 million people suffer from IBD, causing considerable suffering, with a prevalence of Crohn’s disease (CD) of 201 per 100,000 population, UC is being equally prevalent [46].
Because Crohn’s disease is mainly a chronic inflammation, the inflammatory cytokine productions in intestinal biopsies have been studied in relation to the pathological grade. This research originally shows that TNF-α and IL-1β are significantly increased in endoscopic biopsies, as well as for TNF-α, IL-1β, and IL-6 in the individual culture supernatant of intestinal biopsies, opening a new way of investigation [47]. Thus, the effect of pentoxifylline was investigated on intestinal inflammation in IBD, showing that pentoxifylline downregulates in vitro TNF-α and IL-1β production by PBMCs and by intestinal organ cultures from patients with Crohn’s disease and ulcerative colitis [48]. Because pentoxifylline, acting on TNF-α and IL-1β, is a non-selective PDE inhibitor [49], PDE activities were investigated in the human normal mucosa and inflamed mucosa of patients with Crohn’s disease. These studies revealed an increase in the % of PDE4 activity (from 42% to 72.5 %, up by p < 0.05%) suggesting that the specific PDE4 inhibitor may be effective in Crohn’s disease [50]. In that way, the use of OPC-6535 (tetomilast) was suggested for the treatment of a variety of oxidative inflammatory intestinal disorders with an abnormal mucosal barrier such as inflammatory bowel disease [51]. As a result, tetomilast was developed by OTSUKA (phase III) to investigate the treatment of UC [52]. Furthermore, it has been clearly reported that the PDE4 inhibitor, rolipram, prevents and reduces experimental colitis in mice and suppresses TNF-α levels in colonic tissues [53]. Interestingly, rolipram is shown to be superior to methylprednisolone in preventing late collagen deposition [54].
5. PDE4 and Osteoporosis
Osteoporosis is a common disease characterized by reduced bone mineral density (BMD) and increased risk of fragility fractures, which is usually caused by osteoclasts, whereas osteopenia corresponds to a lower bone density than normal and represents the stage before osteoporosis. By 1970, no medications against osteoporosis were being investigated. At the time, fractures were not even recognized as an illness but were considered part of normal aging [55]. Nonetheless, osteoporosis is a public health issue worldwide, affecting over 200 million people. An estimated 30% to 50% of postmenopausal women have this disease. The prevalence (June 2009 to June 2010) of osteoporosis and osteopenia in healthy active workers is 18% [56]. However, it must be pointed out that osteoporosis is an age-associated disease. In the Eastern Mediterranean region, the overall pooled prevalence of osteoporosis was 24.4%, pointing out the necessity of investigating therapeutic approaches [57].
In that way, a study showed that denbufylline, an inhibitor of PDE4 [58][59], was shown to increase the number of mineralized nodules and decrease the number of osteoclast-like cells, suggesting that it should be a therapeutic drug for bone loss [60]. Therefore, this research was reinforced by studying the effect of a new PDE4 inhibitor, XT-44, on mineralized nodule formation, as well as in vivo in ovariectomized female Wistar rats. XT-44 stimulated the formation of mineralized nodules, while it inhibited the formation of osteoclast-like cells in mouse bone marrow culture. Interestingly, in ovariectomized female rats, 1 mg/kg per os (every 2 days for 8 weeks) increased bone mineral density, indicating that XT-44 may be effective in osteoporosis treatment [61].
6. PDE4 and Malignancies
According to MedlinePlus Medical Encyclopedia, “the malignancy” refers to the presence of cancerous cells that have the ability to spread to other sites in the body (https://medlineplus.gov/enc/article/002253.htm, accessed on 13 November 2020). The global cancer incidence and mortality in 2020 were estimated at 19.3 million, almost a million new cases of cancer and 10 million deaths from cancer. The global cancer burden is expected to be 28.4 million cases in 2040 [62].
The use of PDE4-Is in various developing cancers was first investigated in various cancer cell lines as well as on cancer tissues [63][64][65]. Therefore, it was shown that PDE4 protein and mRNA up-regulations are associated in vivo with human endothelial cell proliferation and angiogenesis [9][66] as well as in vivo cancer development in mice [67]. Similarly, there has been reported a phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulating angiogenesis in B-cell lymphoma [68]. Since the PDE4-I, piclamilast, was able to potentiate the cyto-differentiating action of retinoids in myeloid leukemia cells [69], the researchers investigated PDE activity and expression in retinoic acid-resistant cell lines in acute promyelocytic leukemia and showed an increase in PDE4 activity, accompanied by an increase in PDE4D expression [70]. In another area, studies performed in mice, rats, and human tissue revealed that PDE4B protects colon adenomas and is inactivated by epigenetic silencing in colon cancer [71].
7. PDE4 and Fatty Liver Disease
Fatty liver disease, currently named nonalcoholic fatty liver disease (NAFLD) according to the practice guidance commissioned by the American Association for the Study of Liver Diseases (AASLD), is defined by: (1) evidence of hepatic steatosis (HS), either by imaging or histology, and (2) lack of secondary causes of hepatic fat accumulation such as significant alcohol consumption, long-term use of a steatogenic medication, or monogenic hereditary disorders [72]. The overall global prevalence of NAFLD diagnosed by imaging is about 25.24% (95% CI, 22.10–28.65) [73]. NAFLD and alcoholic liver disease (ALD) are the leading causes of liver-related morbidity and mortality and are important causes of liver transplantation [74]. Using bile-duct ligation, as a cholestatic liver injury model, Gobejishvili and colleagues 2013 [75] demonstrated that induction of hepatic PDE4A, B, and D plays a causal role in the development of liver injury and fibrosis. In addition, roflumilast PDE4-I improved glucose tolerance, reduced insulin resistance, and decreased steatohepatitis in mice, increasing the cellular respiratory capacity of hepatocytes [45]. Interestingly, Vonghia et al., 2019 [74] mentioned that the compound tipelukast, also known as MN-001, designed as a leukotriene receptor antagonist, orally bioavailable, being anti-fibrotic and anti-inflammatory in pre-clinical models, interestingly, inhibits PDE3 and PDE4. However, a lack of efficacy of a PDE4-I in phase 1 and 2 trials of patients with non-alcoholic steatohepatitis has already been shown [76].
Overall, these data suggest that today PDE4-Is have failed in the treatment of NAFLD but have induced interesting positive data regarding ALD treatment. Thus, it has been shown that altered PDE4B plays a critical role in alcohol-induced steatosis [77]. A recent review designs PDE4 inhibition as a therapeutic target for ALD as much as, expression of the PDE4 subfamilies is significantly up-regulated in conjunction with markedly decreased cAMP levels in hepatic tissues of patients with severe ALD [78]. It should be noted that inhibition of PDE4 decreases ethanol intake in mice, extending the contribution of PDE4 to liver diseases [79]. Thus, contrary to ALD, one could speculate that liver PDE4 is not sufficiently induced in NAFLD to demonstrate changes in PDE4 expression and efficiency of PDE4-Is. Nonetheless, overexpression of PDE4 in the mouse liver is sufficient to trigger NAFLD and hypertension and can be avoided and even reversed by roflumilast PDE4-I [80]. A very recent study has supported the clinical use of a novel liposomal rolipram formulation to reduce emesis [81].
8. PDE4 and Depression
Major depression is a common illness that severely limits psychosocial functioning and diminishes quality of life. In 2008, the WHO ranked major depression as the third cause of burden of disease worldwide and projected that the disease will rank first by 2030, the 12-month prevalence of major depressive disorder (MDD) being approximately 6.6%, and the lifetime risk being 15–18% [82].
The potential antidepressant activity of rolipram, as a cAMP phosphodiesterase inhibitor, was canonically demonstrated in mice by Wachtel H, 1983 [83]. In that way, rolipram was first reported as a specific PDE4-I [1]. Rolipram was shown to have antidepressant-like effects on behavior maintained by differential reinforcement of low rat response [84] and to facilitate the establishment of long-lasting long-term potentiation and improve memory [85]. In PDE4D knockout (PDE4D−/−) mice, the loss of PDE4D in the cerebral cortex and hippocampus, interestingly, induces an antidepressant-like effect on behavior and reduces sensitivity to rolipram. Furthermore, rolipram potentiated isoproterenol-induced cyclic AMP formation only in the PDE4D+/+ mice. Interestingly, the PDE4D-regulated cyclic AMP signaling may play a role in the pathophysiology and pharmacotherapy of depression [86].
9. PDE4, and COVID-19
As previously [87] stated, during the 1990s, PDE4 might participate in viral infections, since the PDE4 inhibitor, rolipram, was canonically shown to inhibit human immunodeficiency virus-1 (HIV-1) replication, and to decrease HIV-1 p24 antigen production in acutely HIV-1 infected PBMCs [88]. This rolipram inhibitory effect on HIV-1 replication in Jurkat and primary T-cells induced by T-cell activation has been confirmed and extended to the production of TNF-α, NFκB, and NFAT activation [89]. Beavo and colleagues showed that infection of CD4+ memory T-cells by HIV-1 requires the expression of PDE4, and that rolipram abolishes HIV-1 DNA nuclear import in memory T cells, pointing out the important contribution of PDE4 in viral infection [90]. Roflumilast inhibits respiratory syncytial virus infection in differentiated human bronchial epithelial cells [91]. However, in the human airway smooth muscle infected by rhinovirus, a non-enveloped RNA virus, surprisingly, inhibition of PDE4 did not overcome cytokine induction [92], opening the hypothesis that the viral envelope is a requisite for cytokine induction and PDE4-I effectiveness. Interestingly, it has been reported that cAMP produced in Tregs is implicated in suppressing the activation and expression of the HIV-1 gene in vivo in humanized mice [93]. Since a relationship between HIV-1 and PDE4 was clearly established, one could wonder whether COVID-19 might modify PDE4 regulation.
Altogether, these data reported for COVID-19 suggest the hypothesis that a PDE4 inhibitor might alleviate both viral infection and tissue inflammation induced by SARS-CoV-2 [87].
Interestingly, a paper published in Nature Medicine (2005), demonstrated that angiotensin-converting enzyme 2 (ACE2) plays a strategic role in SARS-CoV-2-induced lung injury. As expected, the injection of SARS-CoV-2 spikes into mice worsens acute lung failure in vivo which can be alleviated by blocking the renin–angiotensin pathway governed by ACE2 [94]. Furthermore, it has been reported that Ang-II, an AT1R agonist, induces PDE4 up-regulation, with a 44% increase in the PDE4A protein, mediating cascade inflammation and oxidative stress. Altogether, this opens a new way of PDE interaction with the renin–angiotensin system [95]. More recently, relationships between PDE4, and cytokine storm were discussed for COVID-19 [96][97][98][99]. In this way, a recent in vitro study (phase III) performed with the inhaled PDE4 inhibitor, tanamilast (CHF6001), showed that it blunts proinflammatory dendritic cell activation by SARS-CoV-2 ssRNAs [100].
References
- Lugnier, C.; Stierlé, A.; Beretz, A.; Schoeffter, P.; Lebec, A.; Wermuth, C.G.; Cazenave, J.P.; Stoclet, J.C. Tissue and substrate specificity of inhibition by alkoxy-aryl-lactams of platelet and arterial smooth muscle cyclic nucleotide phosphodiesterases relationship to pharmacological activity. Biochem. Biophys. Res. Commun. 1983, 113, 954–959.
- Stoclet, J.C.; Keravis, T.; Komas, N.; Lugnier, C. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiovascular diseases. Expert Opin. Investig. Drugs 1995, 4, 1081–1100.
- Komas, N.; Lugnier, C.; Stoclet, J.C. Endothelium-dependent and independent relaxation of the rat aorta by cyclic nucleotide phosphodiesterase inhibitors. Br. J. Pharmacol. 1991, 104, 495–503.
- Lugnier, C.; Komas, N. Modulation of vascular cyclic nucleotide phosphodiesterases by cyclic GMP: Role in vasodilatation. Eur. Heart J. 1993, 14 (Suppl. 1), 141–148.
- Eckly, A.E.; Lugnier, C. Role of phosphodiesterases III and IV in the modulation of vascular cyclic AMP content by the NO/cyclic GMP pathway. Br. J. Pharmacol. 1994, 113, 445–450.
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily and smooth muscle signaling. In New Frontiers in Smooth Muscle Biology and Physiology; Savineau, J.P., Ed.; Transworld Research, Network: Kerala, India, 2007; pp. 269–289.
- Lugnier, C.; Schini, V.B. Characterization of cyclic nucleotide phosphodiesterases from cultured bovine aortic endothelial cells. Biochem. Pharmacol. 1990, 39, 75–84.
- Keravis, T.; Komas, N.; Lugnier, C. Cyclic nucleotide hydrolysis in bovine aortic endothelial cells in culture: Differential regulation in cobblestone and spindle phenotypes. J. Vasc. Res. 2000, 37, 235–249.
- Favot, L.; Keravis, T.; Lugnier, C. Modulation of VEGF-induced endothelial cell cycle protein expression through cyclic AMP hydrolysis by PDE2 and PDE4. Thromb. Haemost. 2004, 92, 634–645.
- Keravis, T.; Silva, A.P.; Favot, L.; Lugnier, C. Role of PDEs in vascular health and disease: Endothelial PDEs and angiogenesis. In Cyclic Nucleotide Phosphodiesterases in Health and Disease; Beavo, J.A., Francis, S.H., Houslay, M., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 417–439.
- Kessler, T.; Lugnier, C. Rolipram increases cyclic GMP content in L-arginine-treated cultured bovine aortic endothelial cells. Eur. J. Pharmacol. 1995, 290, 163–167.
- Suttorp, N.; Ehreiser, P.; Hippenstiel, S.; Fuhrmann, M.; Krüll, M.; Tenor, H.; Schudt, C. Hyperpermeability of pulmonary endothelial monolayer: Protective role of phosphodiesterase isoenzymes 3 and 4. Lung 1996, 174, 181–194.
- Rampersad, S.N.; Ovens, J.D.; Huston, E.; Umana, M.B.; Wilson, L.S.; Netherton, S.J.; Lynch, M.J.; Baillie, G.S.; Houslay, M.D.; Maurice, D.H. Cyclic AMP phosphodiesterase 4D (PDE4D) tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J. Biol. Chem. 2010, 285, 33614–33622.
- Hubert, F.; Belacel-Ouari, M.; Manoury, B.; Zhai, K.; Domergue-Dupont, V.; Mateo, P.; Joubert, F.; Fischmeister, R.; Leblais, V. Alteration of vascular reactivity in heart failure: Role of phosphodiesterases 3 and 4. Br. J. Pharmacol. 2014, 171, 5361–5375.
- Varona, S.; Puertas, L.; Galán, M.; Orriols, M.; Cañes, L.; Aguiló, S.; Camacho, M.; Sirvent, M.; Andrés, V.; Martínez-González, J.; et al. Rolipram prevents the formation of abdominal aortic aneurysm (AAA) in Mice: PDE4B as a target in AAA. Antioxidants 2021, 10, 460.
- Wollborn, J.; Siemering, S.; Steiger, C.; Buerkle, H.; Goebel, U.; Schick, M.A. Phosphodiesterase-4 inhibition reduces ECLS-induced vascular permeability and improves microcirculation in a rodent model of extracorporeal resuscitation. Am. J. Physiol. Heart. Circ. Physiol. 2019, 316, H751–H761.
- Tsien, R.W. Cyclic AMP and contractile activity in heart. Adv. Cycl. Nucleotide Res. 1977, 8, 363–420.
- Harrison, S.A.; Reifsnyder, D.H.; Gallis, B.; Cadd, G.G.; Beavo, J.A. Isolation and characterization of bovine cardiac muscle cGMP- inhibited phosphodiesterase: A receptor for new cardiotonic drugs. Mol. Pharmacol. 1986, 29, 506–514.
- Komas, N.; Lugnier, C.; Le Bec, A.; Serradeil-Le Gal, C.; Barthelemy, G.; Stoclet, J.C. Differential sensitivity to cardiotonic drugs of cyclic AMP phosphodiesterases isolated from canine ventricular and sinoatrial-enriched tissues. J. Cardiovasc. Pharmacol. 1989, 14, 213–220.
- Muller, B.; Lugnier, C.; Stoclet, J.C. Implication of cyclic AMP in the positive inotropic effects of cyclic GMP-inhibited cyclic AMP phosphodiesterase inhibitors on guinea pig isolated left atria. J. Cardiovasc. Pharmacol. 1990, 15, 444–451.
- Lanfear, D.E.; Hasan, R.; Gupta, R.C.; Williams, C.; Czerska, B.; Tita, C.; Bazari, R.; Sabbah, H.N. Short term effects of milrinone on biomarkers of necrosis, apoptosis, and inflammation in patients with severe heart failure. J. Transl. Med. 2009, 7, 67.
- Prigent, A.F.; Fougier, S.; Nemoz, G.; Anker, G.; Pacheco, H.; Lugnier, C.; Lebec, A.; Stoclet, J.C. Comparison of cyclic nucleotide phosphodiesterase isoforms from rat heart and bovine aorta. Separation and inhibition by selective reference phosphodiesterase inhibitors. Biochem. Pharmacol. 1988, 37, 3671–3681.
- Stoclet, J.C.; Boulanger-Saunier, C.; Lassègue, B.; Lugnier, C. Cyclic nucleotides and calcium regulation in heart and smooth muscle cells. Ann. N. Y. Acad. Sci. 1988, 522, 106–115.
- Lugnier, C.; Gauthier, C.; Le Bec, A.; Soustre, H. Cyclic nucleotide phosphodiesterases from frog atrial fibers: Isolation and drug sensitivities. Am. J. Physiol. 1992, 262, H654–H660.
- Muller, B.; Stoclet, J.C.; Lugnier, C. Cytosolic and membrane-bound cyclic nucleotide phosphodiesterases from guinea pig cardiac ventricles. Eur. J. Pharmacol. 1992, 225, 263–272.
- Muller, B.; Lugnier, C.; Stoclet, J.C. Involvement of rolipram-sensitive cyclic AMP phosphodiesterase in the regulation of cardiac contraction. Cardiovasc. Pharmacol. 1990, 16, 796–803.
- Eschenhagen, T. PDE4 in the human heart–major player or little helper? Br. J. Pharmacol. 2013, 168, 524–527.
- Lugnier, C.; Muller, B.; Le Bec, A.; Beaudry, C.; Rousseau, E. Characterization of indolidan- and rolipram-sensitive cyclic nucleotide phosphodiesterases in canine and human cardiac microsomal fractions. J. Pharmacol. Exp. Ther. 1993, 265, 1142–1151.
- Lugnier, C.; Keravis, T.; Le Bec, A.; Pauvert, O.; Proteau, S.; Rousseau, E. Characterization of cyclic nucleotide phosphodiesterase isoforms associated to isolated cardiac nuclei. Biochim. Biophys. Acta 1999, 1472, 431–446.
- Bedioune, I.; Lefebvre, F.; Lechêne, P.; Varin, A.; Domergue, V.; Kapiloff, M.S.; Fischmeister, R.; Vandecasteele, G. PDE4 and mAKAPβ are nodal organizers of β2-ARs nuclear PKA signalling in cardiac myocytes. Cardiovasc. Res. 2018, 114, 1499–1511.
- Verde, I.; Pahlke, G.; Salanova, M.; Zhang, G.; Wang, S.; Coletti, D.; Onuffer, J.; Jin, S.L.; Conti, M. Myomegalin is a novel protein of the Golgi/centrosome that interacts with a cyclic nucleotide phosphodiesterase. J. Biol. Chem. 2001, 276, 11189–11198.
- Levian, C.; Ruiz, E.; Yang, X. The pathogenesis of obesity from a genomic and systems biology perspective. Yale J. Biol. Med. 2014, 87, 113–126.
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Briefs 2020, 360, 1–8.
- Chung, J.H.; Manganiello, V.; Dyck, J.R. Resveratrol as a calorie restriction mimetic: Therapeutic implications. Trends Cell Biol. 2012, 22, 546–554.
- Omar, B.; Banke, E.; Ekelund, M.; Frederiksen, S.; Degerman, E. Alterations in cyclic nucleotide phosphodiesterase activities in omental and subcutaneous adipose tissues in human obesity. Nutr. Diabetes 2011, 1, e13.
- Kraynik, S.M.; Miyaoka, R.S.; Beavo, J.A. PDE3 and PDE4 isozyme-selective inhibitors are both required for synergistic activation of brown adipose tissue. Mol. Pharmacol. 2013, 83, 1155–1165.
- Jensterle, M.; Salamun, V.; Kocjan, T.; Vrtacnik Bokal, E.; Janez, A. Short term monotherapy with GLP-1 receptor agonist liraglutide or PDE 4 inhibitor roflumilast is superior to metformin in weight loss in obese PCOS women: A pilot randomized study. J. Ovarian Res. 2015, 8, 32.
- Zhang, R.; Maratos-Flier, E.; Flier, J.S. Reduced adiposity and high-fat diet-induced adipose inflammation in mice deficient for phosphodiesterase 4B. Endocrinology 2008, 150, 3076–3082.
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141.
- Yazdanpanah, S.; Rabiee, M.; Tahriri, M.; Abdolrahim, M.; Rajab, A.; Jazayeri, H.E.; Tayebi, L. Evaluation of glycated albumin (GA) and GA/HbA1c ratio for diagnosis of diabetes and glycemic control: A comprehensive review. Crit. Rev. Clin. Lab. Sci. 2017, 54, 219–232.
- Lugnier, C. Cyclic nucleotide phosphodiesterase families in intracellular signaling and diabetes. Adv. Exp. Med. Biol. 2001, 498, 253–261.
- Parker, J.C.; VanVolkenburg, M.A.; Ketchum, R.J.; Brayman, K.L.; Andrews, K.M. Cyclic AMP phosphodiesterases of human and rat islets of Langerhans: Contributions of types III and IV to the modulation of insulin secretion. Biochem. Biophys. Res. Commun. 1995, 217, 916–923.
- Waddleton, D.; Wu, W.; Feng, Y.; Thompson, C.; Wu, M.; Zhou, Y.P.; Howard, A.; Thornberry, N.; Li, J.; Mancini, J.A. Phosphodiesterase 3 and 4 comprise the major cAMP metabolizing enzymes responsible for insulin secretion in INS-1 (832/13) cells and rat islets. Biochem. Pharmacol. 2008, 76, 884–893.
- Tian, G.; Sågetorp, J.; Xu, Y.; Shuai, H.; Degerman, E.; Tengholm, A. Role of phosphodiesterases in the shaping of sub-plasma-membrane cAMP oscillations and pulsatile insulin secretion. J. Cell Sci. 2012, 125, 5084–5095.
- Möllmann, J.; Kahles, F.; Lebherz, C.; Kappel, B.; Baeck, C.; Tacke, F.; Werner, C.; Federici, M.; Marx, N.; Lehrke, M. The PDE4 inhibitor roflumilast reduces weight gain by increasing energy expenditure and leads to improved glucose metabolism. Diabetes Obes. Metab. 2017, 19, 496–508.
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Jana, G.; Hashash, J.G. A comprehensive review and update on Crohn’s disease. Dis. Mon. 2018, 64, 20–57.
- Reimund, J.-M.; Wittersheim, C.; Dumont, S.; Muller, C.D.; Kenney, J.S.; Baumann, R.; Poindron, P.; Duclos, B. Increased production of tumour necrosis factor-α, interleukin-1β, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn’s disease. Gut 1996, 39, 684–689.
- Reimund, J.M.; Dumont, S.; Muller, C.D.; Kenney, J.S.; Kedinger, M.; Baumann, R.; Poindron, P.; Duclos, B. In vitro effects of oxpentifylline on inflammatory cytokine release in patients with inflammatory bowel disease. Gut 1997, 40, 475–480.
- Tetsi, L.; Charles, A.L.; Paradis, S.; Lejay, A.; Talha, S.; Geny, B.; Lugnier, C. Effects of cyclic nucleotide phosphodiesterases (PDEs) on mitochondrial skeletal muscle functions. Cell. Mol. Life Sci. 2017, 74, 1883–1893.
- Arondel, Y.; Keravis, T.; Le Bec, A.; Baumann, R.; Duclos, B.; Reimund, J.M.; Lugnier, C. First characterisation of cyclic nucleotide phosphodiesterase isoforms in normal human mucosa and inflamed mucosa from Crohn’s disease patients. Gastroenterology 1999, 116, G3716.
- Banan, A.; Fitzpatrick, L.; Zhang, Y.; Keshavarzian, A. OPC-compounds prevent oxidant-induced carbonylation and depolymerization of the F-actin cytoskeleton and intestinal barrier hyperpermeability. Free Radic. Biol. Med. 2001, 30, 287–298.
- O’Mahony, S. Tetomilast. IDrugs 2005, 8, 502–507.
- Hartmann, G.; Bidlingmaier, C.; Siegmund, B.; Albrich, S.; Schulze, J.; Tschoep, K.; Eigler, A.; Lehr, H.A.; Endres, S. Specific type IV phosphodiesterase inhibitor rolipram mitigates experimental colitis in mice. J. Pharmacol. Exp. Ther. 2000, 292, 22–30.
- Videla, S.; Vilaseca, J.; Medina, C.; Mourelle, M.; Guarner, F.; Salas, A.; Malagelada, J.R. Selective inhibition of phosphodiesterase-4 ameliorates chronic colitis and prevents intestinal fibrosis. J. Pharmacol. Exp. Ther. 2006, 316, 940–945.
- Gallagher, J.C. Advances in osteoporosis from 1970 to 2018. Menopause 2018, 25, 1403–1417.
- Barrios-Moyano, A.; De la Peña-García, C. Prevalencia de osteoporosis y osteopenia en pacientes laboralmente activos. Acta Ortop. Mex. 2018, 32, 131–133.
- Zamani, M.; Zamani, V.; Heidari, B.; Parsian, H.; Esmaeilnejad-Ganji, S.M. Prevalence of osteoporosis with the World Health Organization diagnostic criteria in the Eastern Mediterranean Region: A systematic review and meta-analysis. Arch. Osteoporos. 2018, 13, 129.
- Lugnier, C.; Stoclet, J.C.; Wilke, R. Phosphodiesterase isoenzymes in different tissues and the selective inhibition by denbufylline. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1989, 339, R106.
- Nicholson, C.D.; Jackman, S.A.; Wilke, R. The ability of denbufylline to inhibit cyclic nucleotide phosphodiesterase and its affinity for adenosine receptors and the adenosine re-uptake site. Br. J. Pharmacol. 1989, 97, 889–897.
- Miyamoto, K.; Waki, Y.; Horita, T.; Kasugai, S.; Ohya, K. Reduction of bone loss by denbufylline, an inhibitor of phosphodiesterase 4. Biochem. Pharmacol. 1997, 54, 613–617.
- Waki, Y.; Horita, T.; Miyamoto, K.; Ohya, K.; Kasugai, S. Effects of XT-44, a phosphodiesterase 4 inhibitor, in osteoblastgenesis and osteoclastgenesis in culture and its therapeutic effects in rat osteopenia models. Jpn. J. Pharmacol. 1999, 79, 477–483.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Kim, D.H.; Lerner, A. Type 4 cyclic adenosine monophosphate phosphodiesterase as a therapeutic target in chronic lymphocytic leukemia. Blood 1998, 92, 2484–2494.
- Marko, D.; Romanakis, K.; Zankl, H.; Fürstenberger, G.; Steinbauer, B.; Eisenbrand, G. Induction of apoptosis by an inhibitor of cAMP-specific PDE in malignant murine carcinoma cells overexpressing PDE activity in comparison to their nonmalignant counterparts. Cell. Biochem. Biophys. 1998, 28, 75–101.
- Narita, M.; Murata, T.; Shimizu, K.; Sugiyama, T.; Nakagawa, T.; Manganiello, V.C.; Tagawa, T. Phosphodiesterase 4 in osteoblastic osteosarcoma cells as a potential target for growth inhibition. Anticancer. Drugs 2003, 14, 377–381.
- Favot, L.; Keravis, T.; Holl, V.; Le Bec, A.; Lugnier, C. VEGF-induced HUVEC migration and proliferation are decreased by PDE2 and PDE4 inhibitors. Thromb. Haemost. 2003, 90, 334–343.
- Abusnina, A.; Keravis, T.; Zhou, Q.; Justiniano, H.; Lobstein, A.; Lugnier, C. Tumour growth inhibition and anti-angiogenic effects using curcumin correspond to combined PDE2 and PDE4 inhibition. Thromb. Haemost. 2015, 113, 319–328.
- Suhasini, A.N.; Wang, L.; Holder, K.N.; Lin, A.P.; Bhatnagar, H.; Kim, S.W.; Moritz, A.W.; Aguiar, R.C.T. A phosphodiesterase 4B-dependent interplay between tumor cells and the microenvironment regulates angiogenesis in B-cell lymphoma. Leukemia 2016, 30, 617–626.
- Parrella, E.; Gianni, M.; Cecconi, V.; Nigro, E.; Barzago, M.M.; Rambaldi, A.; Rochette-Egly, C.; Terao, M.; Garattini, E. Phosphodiesterase IV inhibition by piclamilast potentiates the cytodifferentiating action of retinoids in myeloid leukemia cells. Cross-talk between the cAMP and the retinoic acid signaling pathways. J. Biol. Chem. 2004, 279, 42026–42040.
- Keravis, T.; Justiniano, H.; Guillemin, M.C.; de Thé, H.; Rochette-Egly, C.; Lugnier, C. cAMP-PDE activity, PDE4 activity and PDE4D protein expression are increased in RA-resistant NB4-R2 cells. In Proceedings of the EMBO Retinoids 2011: Mechanisms, Biology and pathology of Signaling by Retinoic acid and Retinoic acid Receptors, Strasbourg, France, 22–25 September 2011.Poster.
- Pleiman, J.K.; Irving, A.A.; Wang, Z.; Toraason, E.; Clipson, L.; Dove, W.F.; Dustin, A.; Deming, D.A.; Newton, M.A. The conserved protective cyclic AMP-phosphodiesterase function PDE4B is expressed in the adenoma and adjacent normal colonic epithelium of mammals and silenced in colorectal cancer. PLoS Genet. 2018, 14, e1007611.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Vonghia, L.; Van Herck, M.A.; Weyler, J.; Francque, S. Targeting myeloid-derived cells: New frontiers in the treatment of non-alcoholic and alcoholic liver disease. Front. Immunol. 2019, 10, 563.
- Gobejishvili, L.; Barve, S.; Breitkopf-Heinlein, K.; Li, Y.; Zhang, J.; Avila, D.V.; Dooley, S.; McClain, C.J. Rolipram attenuates bile duct ligation-induced liver injury in rats: A potential pathogenic role of PDE4. J. Pharmacol. Exp. Pathol. 2013, 347, 80–90.
- Ratziu, V.; Bedossa, P.; Francque, S.M.; Larrey, D.; Aithal, G.P.; Serfaty, L.; Voiculescu, M.; Preotescu, L.; Nevens, F.; De Lédinghen, V.; et al. Lack of efficacy of an inhibitor of PDE4 in phase 1 and 2 trials of patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2014, 12, 1724–1730.e5.
- Avila, D.V.; Barker, D.F.; Zhang, J.; McClain, C.J.; Barve, S.; Gobejishvili, L. Dysregulation of hepatic cAMP levels via altered Pde4b expression plays a critical role in alcohol-induced steatosis. J. Pathol. 2016, 240, 96–107.
- Rodriguez, W.E.; Wahlang, B.; Wang, Y.; Zhang, J.; Vadhanam, M.V.; Joshi-Barve, S.; Bauer, P.; Cannon, R.; Ahmadi, A.R.; Sun, Z.; et al. Phosphodiesterase 4 inhibition as a therapeutic target for alcoholic liver disease: From bedside to bench. Hepatology 2019, 70, 1958–1971.
- Hu, W.; Lu, T.; Chen, A.; Huang, Y.; Hansen, R.; Chandler, L.J.; Zhang, H.-T. Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology 2011, 218, 331–339.
- Tao, X.; He, H.; Peng, J.; Xu, R.; Fu, J.; Hu, Y.; Li, L.; Yang, X.; Feng, X.; Zhang, C.; et al. Overexpression of PDE4D in mouse liver is sufficient to trigger NAFLD and hypertension in a CD36-TGF-beta1 pathway: Therapeutic role of roflumilast. Pharmacol. Res. 2022, 175, 106004.
- Gobejishvili, L.; Rodriguez, W.E.; Bauer, P.; Wang, Y.; Soni, C.; Lydic, T.; Barve, S.; McClain, C.; Maldonado, C. Novel liposomal rolipram formulation for clinical application to reduce emesis. Drug Des. Devel. Ther. 2022, 16, 1301–1309.
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312.
- Wachtel, H. Potential antidepressant activity of rolipram and other selective cyclic adenosine 3′,5′-monophosphate phosphodiesterase inhibitors. Neuropharmacology 1983, 22, 267–272.
- O’Donnell, J.M. Antidepressant-like effects of rolipram and other inhibitors of cyclic adenosine monophosphate phosphodiesterase on behavior maintained by differential reinforcement of low response rate. J. Pharmacol. Exp. Ther. 1993, 264, 1168–1178.
- Barad, M.; Bourtchouladze, R.; Winder, D.G.; Golan, H.; Kandel, E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc. Natl. Acad. Sci. USA 1998, 95, e15020–e15025.
- Zhang, H.T.; Huang, Y.; Jin, S.L.; Frith, S.A.; Suvarna, N.; Conti, M.; O’Donnell, J.M. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology 2002, 27, 587–595.
- Lugnier, C.; Al-Kuraishy, H.M.; Rousseau, E. PDE4 inhibition as a therapeutic strategy for improvement of pulmonary dysfunctions in Covid-19 and cigarette smoking. Biochem. Pharmacol. 2021, 185, 114431.
- Angel, J.B.; Saget, B.M.; Walsh, S.P.; Greten, T.F.; Dinarello, C.A.; Skolnik, P.R.; Endres, S. Rolipram, a specific type IV phosphodiesterase inhibitor, is a potent inhibitor of HIV-1 replication. AIDS 1995, 9, 1137–1144.
- Navarro, J.; Punzon, C.; Jimenez, J.L.; Fernandez-Cruz, E.; Pizarro, A.; Fresno, M.; Muñoz-Fernández, M.A. Inhibition of phosphodiesterase type IV suppresses human immunodeficiency virus type 1 replication and cytokine production in primary T cells: Involvement of NF-κB and NFAT. J. Virol. 1998, 72, 4712–4720.
- Sun, Y.; Li, L.; Lau, F.; Beavo, J.A.; Clark, E.A. Infection of CD4+ memory T cells by HIV-1 requires expression of phosphodiesterase 4. J. Immunol. 2000, 165, 1755–1761.
- Mata, M.; Martinez, I.; Melero, J.A.; Tenor, H.; Cortijo, J. Roflumilast inhibits respiratory syncytial virus infection in human differentiated bronchial epithelial cells. PLoS ONE 2013, 8, e69670.
- Van Ly, D.; De Pedro, M.; James, P.; Morgan, L.; Black, J.L.; Burgess, J.K.; Oliver, B.G. Inhibition of phosphodiesterase 4 modulates cytokine induction from toll like receptor activated, but not rhinovirus infected, primary human airway smooth muscle. Respir. Res. 2013, 14, 127.
- Li, G.; Nunoya, J.I.; Cheng, L.; Reszka-Blanco, N.; Tsao, L.C.; Jeffrey, J.; Su, L. Regulatory T Cells Contribute to HIV-1 Reservoir persistence in CD4+ T cells through cyclic adenosine monophosphate–dependent mechanisms in humanized mice in vivo. J. Inf. Dis. 2017, 216, 1579–1591.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Feng Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS Coronavirus-Induced lung injury. Nat. Med. 2005, 11, 875–879.
- Mokni, W.; Keravis, T.; Etienne-Selloum, N.; Walter, A.; Kane, M.O.; Schini-Kerth, V.B.; Lugnier, C. Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PLoS ONE 2010, 5, e14227.
- Dalamaga, M.; Karampela, I.; Mantzoros, C.S. Commentary: Phosphodiesterase 4 inhibitors as potential adjunct treatment targeting the cytokine storm in COVID-19. Metabolism 2020, 109, 154282.
- Chemboli, R.; Kapavarapu, R.; Deepti, K.; Prasad, K.R.S.; Reddy, A.G.; Kumar, A.V.D.N.; Rao, M.V.B.; Pal, M. Pyrroloquinoxalines in attenuating cytokine storm in COVID-19: Their sonochemical synthesis and in silico/in vitro assessment. J. Mol. Struct. 2021, 1230, 129868.
- Bridgewood, C.; Damiani, G.; Sharif, K.; Watad, A.; Bragazzi, N.L.; Quartuccio, L.; Savic, S.; McGonagle, D. Rationale for evaluating PDE4 inhibition for mitigating against severe inflammation in COVID-19 pneumonia and beyond. Isr. Med. Assoc. J. 2020, 22, 335–339.
- Sugin Lal Jabaris, S.; Ranju, V. Scope of adjuvant therapy using roflumilast, a PDE-4 inhibitor against COVID-19. Pulm. Pharmacol. Ther. 2021, 66, 101978.
- Nguyen, H.O.; Schioppa, T.; Tiberio, L.; Facchinetti, F.; Villetti, G.; Civelli, M.; Del Prete, A.; Sozio, F.; Gaudenzi, C.; Passari, M.; et al. The PDE4 Inhibitor Tanimilast Blunts Proinflammatory Dendritic Cell Activation by SARS-CoV-2 ssRNAs. Front. Immunol. 2022, 12, 797390.
More