Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 3 by Dean Liu.
One of the main sources of non-ferrous and precious metals is sulfide ores. The study results of the surface redox transformations of minerals, galvanic effect, cathodic oxygen reduction reaction on the surface of sulfides are presented. The electrochemical properties of sulfide minerals are manifested both in the industrial processes of flotation and hydrometallurgy and in the natural geological setting or during the storage of sulfide-containing mining, mineral processing, and metallurgical industry waste.
- electrochemistry of sulfides
- flotation
- hydrometallurgy
1. Introduction
Sulfide minerals are an important group of ore minerals and a major source of non-ferrous and precious metals. Sulfide minerals are known for high conductivity, which determines the abrupt change in the potential at the sulfide–solution interface [1][2][3][4][5][6][7]. Sulfides such as pentlandite ((Fe,Ni)9S8), covellite (CuS), galena (PbS), and pyrrhotite (Fe1-xS) are close to metals in terms of conductivity. Semiconductor minerals with good conductivity include pyrite (FeS2), bornite (Cu5FeS4), chalcosine (Cu2S), chalcopyrite (CuFeS2), arsenopyrite (FeAsS) [5].
A relevant research and practical task is to improve the contrast of the physicochemical and process properties of sulfide minerals to achieve high separation performance in ore concentration circuits. Multiple existing studies convincingly show that the sorption mechanism of the collector, sulfides flotation kinetics, and the interactions between the minerals in intergrowths have an electrochemical background [1][2][3][4][5][6][7][8]. Pioneering studies in this field include those by Plaksin et al. [1][2][3]. A comprehensive study of the process of interaction of sulfhydryl reagents with sulfides made it possible to discover a number of fundamentally new patterns in the mechanism of xanthate sorption on sulfide minerals. The effect of the electrochemical properties of minerals and the concentration of free electron vacancies on the distribution of the collector and the ratio of xanthate to dixanthogen on sulfide surfaces was experimentally established. By manipulating the state of the mineral surfaces through various physical and chemical impacts, it is possible to modify their electrochemical properties and, thereby, control the flotation of sulfides [1][2][3][4][5][6][7][8][9][10].
Currently, for the processing of low-grade sulfide ores and technogenic mineral resources (overburden, concentration tailings, and smelter slags), heap and bioleaching methods are increasingly being used [11]. The electrochemical properties of sulfides have a decisive influence on the leaching kinetics, the concentration of non-ferrous metals in pregnant solutions, and recovery [12][13][14][15][16]. It should be noted that the utilization of operando/in situ techniques, e.g., X-ray absorption and Raman spectroscopy, can more accurately track the phase, composition, local atomic coordination of sulfides during electrochemistry process.
In addition to process engineering challenges, the environmental aspect should also be taken into consideration (Figure 1).
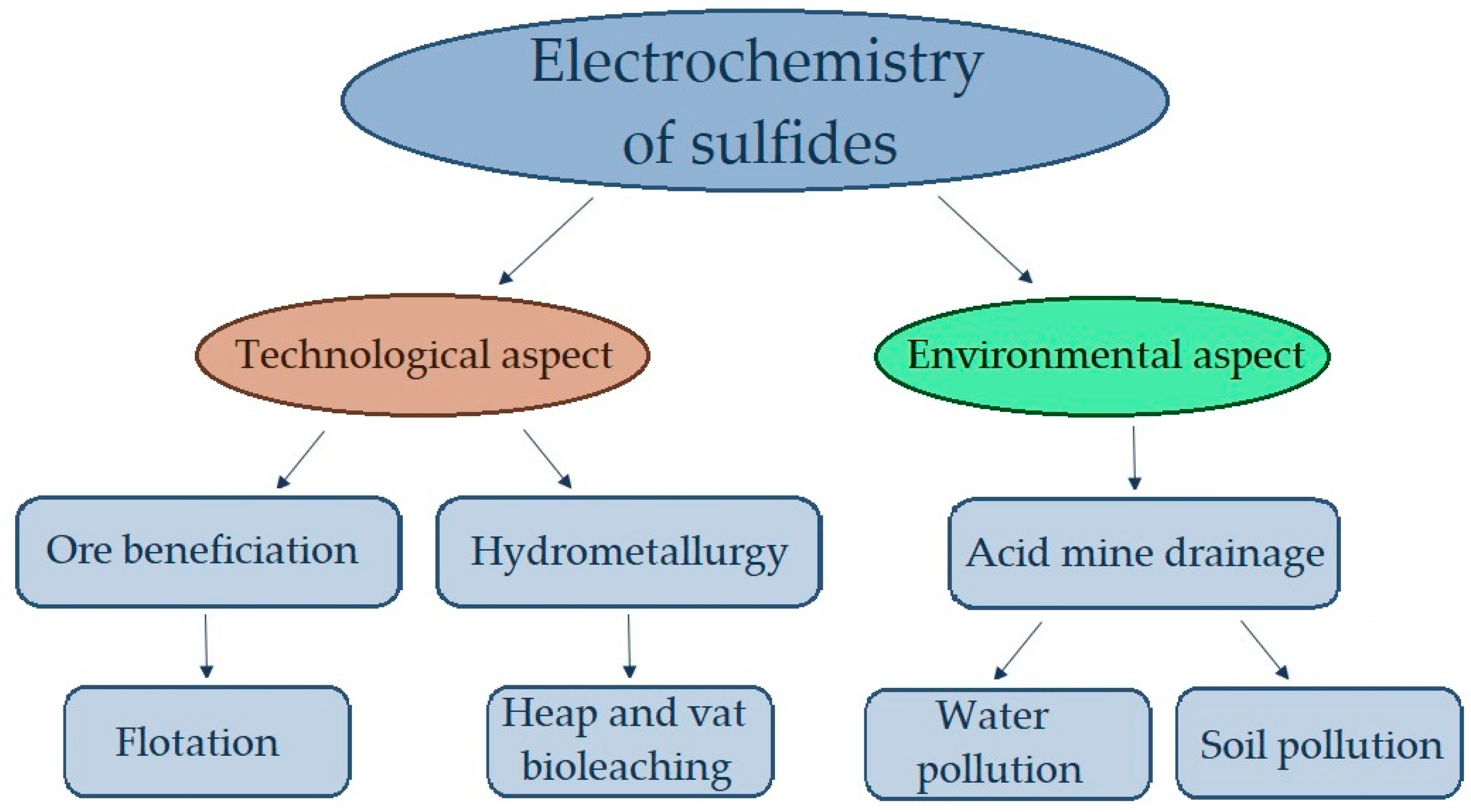
Figure 1. Technological and environmental aspects of the electrochemistry of sulfides.
Sulfide-containing ore mining, mineral processing, and metallurgical wastes are known to be some of the most environmentally hazardous types of industrial waste [17][18]. During waste storage, sulfide minerals, in particular pyrite and pyrrhotite, oxidize forming sulfuric acid (acid mine drainage (AMD)), ferrous and non-ferrous sulfates. Chemical weathering of tailings proceeds much faster than in a natural geological environment. Oxidative dissolution of sulfides as a result of galvanic interactions is one of the most important agents of environmental pollution [17][19][20]. To better understand the nature and mechanisms of chemical weathering, studies of the physicochemical properties of minerals, including electrochemical properties, are needed.
ReThisearchers paper presents a brief review of the existing literature on the electrochemical behavior of some common industrially important sulfide minerals in aqueous solutions and the process of oxygen reduction on the surface of sulfide minerals, which ensures the occurrence of processes important for the processing of sulfide ores (oxidation and leaching reactions, chemisorption and oxidation of sulfhydryl collectors) and in processes weathering occurring in sulfide-containing wastes of mining enterprises. The results of studies of galvanic interactions of sulfides in technological processes—flotation and hydrometallurgy—and environmental aspects of studying the electrochemical properties of sulfide minerals, which must be taken into account to predict the processes of AMD generation and the transfer of heavy metals and other toxic elements to soil and surface water during the storage of mining waste, are also reflected.
2. Electrochemistry of Sulfides in Aqueous Solutions
2.1. Iron Sulfides
Iron sulfides are represented by such minerals as pyrite and marcasite (FeS2), greigite (Fe3S4), pyrrhotite (Fe1xS), mackinawite (FeS1-x), and troilite (FeS). Pyrite and pyrrhotite are the most abundant minerals found in polymetallic ores.
The electrochemical properties of pyrite and pyrrhotite are presented in great detail in [21][22][23][24][25][26][27]. Cathodic and anodic decomposition of pyrite and pyrrhotite in acidic environments is discussed in [22][24][26] without consideration of intermediate reactions preceding decomposition processes. Surface redox transformations of pyrite and pyrrhotite in the stability region of minerals in a wide pH range were studied by cyclic voltammetry in [21]. The authors note that the surface of iron sulfide minerals, depending on pH, is covered with either sulfur or sulfur and iron(III) hydroxide; this film is quite porous and does not inhibit electrochemical dissolution. However, the anodic processes on sulfide electrodes as described in [21] mainly concern the electrochemical oxidation of the products of the preliminary cathodic polarization of these minerals.
Electrochemical oxidation of pyrite in 1 M of HCl was studied in [28] using a wide range of methods. It was found that oxidation includes a series of sequential and parallel reaction stages, as a result of which the
Fe2+ → Fe3+,
S → S2O32− → HSO4−
The author considers the effect of a borate electrolyte (a Na2B4O7 solution) on the kinetics of the electrochemical oxidation of pyrite in [29]. The borate electrolyte was used in most studies as a model buffer system inert with respect to minerals. According to the results obtained, the first stage of the interaction is the irreversible one-electron reaction of formation of a FeS2···[B(OH)4]ads. complex.
Fourier-transform infrared spectroscopy (FTIR) spectra of the pyrite electrode–electrolyte interface were collected in situ and interpreted in [30] at a polarization of −0.5 to +0.9 V relative to the standard hydrogen electrode (SHE) and pH 9.2. An electrochemical model of pyrite oxidation in aqueous environments was proposed. The cathodic half-reaction proceeds in sulfur-deficient regions of pyrite or on FeS defects. The anodic half-reaction proceeds along the thiosulfate path in the areas of “ordinary” pyrite, due to the Fermi level in the upper part of the valence band. The main role of the oxidizing agent is to maintain a high potential on the anode sites. Direct (chemical) oxidation of pyrite is an indirect effect and consists in the interaction of the oxidizing agent with the products of the anodic reaction.
The effect of polysulfide formation on the kinetics of pyrite dissolution in a 1 M HCl solution was examined in [31]. Pyrite was oxidized electrochemically at various anodic dissolution potentials. Further, the thickness of the polysulfide layer was estimated by two methods: using X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. Both methods showed that the thickness of the polysulfide layer increases with increasing potential. At the same time, the current increases exponentially with increasing potential, which indicates the absence of the passivating effect of polysulfides.
Electrochemical properties of pyrrhotite were studied in the works by Chanturiya and Wigdergauz, Buckley, Woods, and other researchers [5][21][24]. The electrochemical behavior of natural pyrrhotite was studied in solutions of 1 M HCl and 0.05 M Na2B4O7 using cyclic voltammetry and potentiostatic method [32]. It was found that the formation of a massive nonequilibrium metal-deficient layer on the mineral surface occurs in 1 M HCl in the potential range from 0.08 to 0 V relative to the silver–silver chloride reference electrode. In this case, the dissolution rate changes by about two orders of magnitude. On the cyclic voltammograms of pyrrhotite with a previously formed metal-deficient layer, an intense cathodic peak is recorded at −0.2 V, and several anodic waves are observed when the potential is reversed. It was found that the cathode peak corresponds to the reductive cleavage of polysulfide with the formation of monosulfide S02− centers, which dissolve in acid to release H2S. Three anodic peaks correspond to the association of S02- with each other and with polysulfide clusters, oxidation of terminal sulfur atoms (S1−) with the association of chains into larger ones and the oxidation of residual S1−. The behavior of an electrode with a metal-deficient layer previously formed in hydrochloric acid and transferred to a borate solution is consistent with these suggestions. In the reactions of pyrrhotite without preliminary acid etching, the same centers participate at lower concentrations. Similar results were obtained by Mikhlin et al. in their study of the electrochemical oxidation of pyrrhotite in a sulfuric acid solution [33][34].
2.2. Copper Sulfides
Industrial minerals include chalcocite (Cu2S), covellite (CuS), chalcopyrite (CuFeS2), to a lesser extent bornite (Cu5FeS4) and cubanite (CuFe2S3). In recent years, a number of studies were conducted on the electrochemistry of a relatively rare sulfoarsenide mineral—enargite (Cu3AsS4).
As noted by Chanturiya and Wigdergauz [5], for copper sulfides, electroleaching processes—cathodic and anodic decomposition—are studied in more detail. The processes preceding decomposition are understood less fully.
One of the early works studied the anodic dissolution of Cu2S in sulfate solutions with simultaneous precipitation of copper [35]. An intermediate product—digenite—was found on the anode surface:
Cu2S → Cu1.8S + 0.2Cu+ + 2e−.
Then, due to a sharp increase in the potential of the electrode, digenite was transformed into covellite. The decomposition of covellite is also superimposed on that of digenite:
CuS → Cu2+ + S + 2e−.
The structure was studied and the phase components of copper(I) sulfide synthesized by the authors were identified [36]. It was shown that crystallization of copper sulfide can lead to the formation of chalcocite (Cu2S) and djurleite phases (Cu31S16), characterized by a deficiency of copper in the crystal lattice and a deviation from the stoichiometric composition. Electrochemical oxidation of the sample in a solution of sulfuric acid was carried out. It was established that at a current density of 1000 A/m2 and a concentration of 100 g/dm3 H2SO4, electrochemical dissolution of copper sulfide proceeds with the formation of a passivating film of sparingly soluble products during the oxidation of copper(I) sulfide to copper(II) sulfide. Moreover, during the oxidation, intermediate non-stoichiometric sulfides were formed in the following sequence:
Cu2S (Cu31S16) → Cu1.8S → Cu1.74S → Cu1.6S → CuS → Cu2+ + S.
The process was accompanied by the transition of copper cations into solution. With the accumulation of elemental sulfur and copper sulfides on the reaction surface of the sample, the rate of electrochemical oxidation decreased due to the difficult removal of products and supply of the reagent to the reaction zone.
The electrochemical behavior of chalcocite in a 0.05 M buffer borate solution (pH 9.2) was studied in [37]. During anodic scanning, the oxidation wave began at −0.1 V relative to the saturated calomel electrode and was accompanied by the transition of Cu2+ ions into solution. During reverse cathodic sweep, a wave of copper sulfide formation was recorded with a peak at ~−0.1 V. Cathodic sweep to more negative potentials led to the emergence of a recovery wave that began at ~−0.58 V with a soluble reduction product HS-. During reverse anode sweep, the HS- ion, apparently interacting with Cu2+, formed copper sulfide with a peak at −0.6 V.
Anodic dissolution of covellite in hydrochloric acid solutions was studied in [38]. It was found that the dissolution rate depends on pH and the concentration of chloride ions. At a Cl− concentration higher than 1 M, the dissolution of CuS is accelerated due to the formation of a CuCl2− complex as a result of the chemical reaction between Cu2+ and CuS.
In papers [39][40][41], the anodic decomposition of chalcopyrite in acidic solutions was discussed. It was shown that at a potential of +0.8 V relative to the silver–silver chloride electrode, selective dissolution of iron occurred with the oxidation of sulfide sulfur to elemental, and at potentials more positive than +1.1 V, sulfide sulfur oxidized to sulfate [37].
Surface redox transformations of chalcopyrite and chalcocite were studied in the region of mineral stability in a wide pH range [42]. Studies performed by cyclic voltammetry included the polarization of the sulfide electrode from the rest potential to the cathode region and then to the anode region. Thus, the electrochemical oxidation of the products of cathodic polarization of chalcopyrite was actually studied.
According to [43], where the stationary polarization curve of chalcopyrite was studied in a 0.1 M Na2B4O7 solution, at a potential greater than −0.044 V relative to a SHE, an iron-deficient layer formed on the surface of chalcopyrite, which passivated the mineral upon reaching a potential of 0.120 V. Transition to the active state occurred at a potential of 0.237 V. At a potential of 0.436 V, chalcopyrite was passivated by metal hydroxides, oxides, and sulfates. At potentials greater than 0.53 V, the mineral dissolved.
Somewhat different results were reported in [44][45]. During the anodic polarization of a mineral in a borate solution, two current maxima were observed, the first of which was associated with the formation of metastable CuS2 and CuFe1-xS2 phases. With a further positive shift of the potential, these phases dissociated with the formation of oxides CuO, Fe2O3, and sulfur.
The reaction of chalcopyrite dissolution in ammoniacal solutions (NH4OH and (NH4)2SO4 at a ratio of 1:1) was studied by potentiostatic and potentiodynamic methods [46]. It was found that the anodic current densities near the mixed potential increased with increasing total ammonia (NH3 + NH4+) concentration from 1 to 3M. It was established that the reaction with respect to ammonia was of the first order. The reaction was of zero order in terms of [OH-] at pH 9 to 10. Tafel equations showed that the average charge transfer coefficient was 0.61. The reaction was controlled by a one-electron charge transfer step. That formed an intermediate copper-depleted layer, which was rapidly oxidized in subsequent electron transfer reactions. The surface precipitate formed during the reaction did not passivate the mineral, but reduced the rate of its dissolution.
The same authors studied the cathodic reduction of copper(II) and oxygen on the surface of chalcopyrite in ammoniacal solutions using cyclic voltammetry and chronoamperometry [47]. It was established that the predominant cathodic reaction during the oxidative dissolution of chalcopyrite in ammonical solutions is the reduction of copper(II). The cathodic reaction rate was found to be inversely proportional to pH and total ammonia concentration. Although tetra-ammine copper(II) ion is the dominant ion, the tri-ammine complex is believed to be the most electrochemically active under these conditions. It was shown that both copper(II) reduction and copper(I) oxidation occur simultaneously at potentials close to the mixed potentials of chalcopyrite in these solutions. The reaction rate is controlled by mass transfer determined by the migration of Cu(I) ions from the surface of the mineral.
Nicol summarized the results of studies of anodic processes on chalcopyrite in alkaline hydroxide and carbonate solutions [48]. Anodic oxidation of chalcopyrite involves active oxidation at low potentials followed by a passivation region and transpassive oxidation at higher potentials. In this case, the formation of solid products of the passivating film does not prevent continued oxidation. The oxidation kinetics is activated with an increase in pH in both hydroxide and carbonate solutions. Coulometric measurements showed that thiosulfate and sulfate ions formed as a result of oxidation in sodium hydroxide solutions. Thiosulfate ion is the main product in carbonate and bicarbonate solutions.
Relatively few studies were focused on the electrochemical behavior of bornite. In one of the earlier papers [49], it was reported that during the anodic oxidation of bornite, an intermediate phase deficient in copper Cu2.5FeS4 formed. It was noted that at temperatures below 50 °C, the anodic dissolution of bornite was controlled by solid-phase diffusion. At temperatures above 65 °C, dissolution was controlled by a chemical reaction.
In a recent article by Chinese researchers, based on the cyclic voltammetry and XPS data, it was shown that the oxidation of bornite proceeds intensively at a potential of more than 0.3 V relative to the silver–silver chloride reference electrode [50]. The main intermediate phases on the surface of bornite during oxidation are elemental sulfur, FeOOH, and CuS.
One of the first studies of the process of electrochemical oxidation of enargite, a valuable but arsenic-containing mineral, usually found in intergrowths with common copper sulfides, was carried out by Ásbjornsson et al. [51]. In a hydrochloric acid solution, they observed a wave in the range from 0.2 to 0.6 V (relative to the SHE) with a maximum at 0.54 V. This wave was associated with the formation of elemental sulfur as a passivating layer. Other parallel reactions with the formation of sulfate ions and arsenic(III) or arsenic(V) were also suggested.
The mechanism of electrochemical oxidation of enargite in sulfuric acid with a concentration of 0.1 mol·L−1 at room temperature was studied in detail by electrochemical and spectroscopic analysis methods [52]. Cyclic voltammetry, potentiodynamic polarization, electrochemical impedance spectroscopy, and the Mott–Schottky method were used. XPS with cold stage, Raman spectroscopy, and synchrotron X-ray absorption near edge structure (XANES) were used to identify product layers on the electrode surface after electrochemical dissolution. A region of passivation of the enargite electrode was found at potentials from 450 to 750 mV relative to the Ag/AgCl electrode. The decrease in the current density in this potential region is due to the formation of a surface layer of As(1-y-z)S4. Copper dissolved more readily than arsenic. The transpassive region with a window between 750 and 900 mV is associated with the partial dissolution of the sulfide sulfur of the passivating surface film to elemental sulfur.
Nicol et al. conducted fundamental studies of the electrochemical behavior of enargite in ammonium sulfate and ammonium chloride solutions in the pH range from 8 to 10 [53] and compared that with the behavior of chalcopyrite under similar conditions. Mixed potential measurements showed that copper(II) is an effective oxidizer of enargite in ammoniacal solutions. Voltammetry confirmed the occurrence of the process of partial passivation of the electrode in the studied solutions. In general, the anodic behavior of enargite in chloride and sulfate solutions is similar, with the main product of anodic oxidation being thiosulfate ion. The relative dissolution rates of chalcopyrite and enargite in ammoniacal solutions were estimated using the mixed potential theory. It was demonstrated that enargite dissolves in such systems faster than chalcopyrite.
Same authors studied cathodic processes [54]. The process of reduction of copper(II) to copper(I) on the surface of enargite in ammonium sulfate solutions proceeds at a high rate and is quasi-reversible. The reaction of the first-order in terms of the concentration of copper(II) ions and its rate is limited by mass transfer even at low overpotentials. The kinetics and mechanism of copper(II) reduction in chloride solutions are quantitatively similar to those occurring in sulfate solutions. The reduction rate decreases with increasing concentrations of ammonia and ammonium ions. According to the developed model, this rate is approximately proportional to the concentration of tri-ammine copper(II) species, whose concentration decreases as the concentrations of ammonia and ammonium ions increase.
2.3. Nickel Sulfides
Pentlandite ((Fe,Ni)9S8) is the main nickel mineral in sulfide copper and nickel deposits. Violarite ((Fe,Ni)3S4) is found in association with pyrrhotite and pentlandite. Nickel also forms several other natural sulfides: millerite (NiS), vaesite (NiS2), heazlewoodite (Ni3S2), polydymite (Ni3S4), and godlevskyite (Ni7S6). These minerals are relatively rare in sulfide ores. At the same time, heazlewoodite is the main component of the copper–nickel concentrate smelting product—feinstein.
The electrochemical behavior of millerite and heazlewoodite was described in detail in [5][55][56][57]. Specifically, the anodic reactions of heazlewoodite are interpreted as mineral transformations Ni3S2 → Ni3-xS2 up to 600 mV relative to the calomel reference electrode; at intermediate potentials of 700–1200 mV, elemental sulfur forms, and at potentials above 1300 mV, the mineral is decomposed with the formation of elemental sulfur and sulfate ion.
In a later paper, Aromaa investigated the anodic dissolution of synthetic heazlewoodite in 1 N sulfuric acid at 25 °C [58]. An analysis of the potentiostatic curves and cyclic voltammograms showed that the formation of intermediate sulfides continued even at high potentials, up to 1600 mV relative to the saturated calomel electrode. It was found that solely sulfur formed up to 900 mV. At a polarization potential of 1000 mV, 90% of elemental sulfur and 10% of sulfates form, at 1600 mV—10% of sulfur, and the rest are sulfate ions.
The electrochemical properties of pentlandite are studied in less detail [59]. In [5], the complex nature and abundance of redox processes on the surface of pentlandite, caused by phase transitions of the mineral, are discussed.
In [60], oxidative dissolution of pentlandite in iron(III) chloride solution was investigated by electrochemical methods, including potentiometry, cyclic voltammetry, galvanostatic polarization, chronopotentiometry, and chronoamperometry. The oxidation products were analyzed using reflected light microscopy, powder X-ray diffraction (XRD), and electron microprobe analysis. It was shown that pentlandite oxidizes directly to elemental sulfur without the formation of any intermediate phases. Violarite formation was not reported. Sulfur layers create obstacles for mass transfer, and the dissolution process is limited by the diffusion of metal atoms in the surface layer of the mineral.
The same authors proposed a similar mechanism for the oxidative dissolution of violarite in an acidified iron(III) chloride solution [61].
In [62], anodic processes on a pentlandite electrode in a carbonate solution were investigated. Anodic polarization of the mineral started at a stationary potential of −0.035 ± 0.010 V relative to Ag/AgCl electrode for various samples of naturally occurring minerals. At an electrode polarization rate of 0.02 V/s, waves were observed at potentials of +0.265 and +0.545 V. The potential of the first wave shifts in the positive direction with an increase in the polarization rate, while in the rate range of 0.02–2 V/s it did not depend on the sweep rate, thus the process was controlled by diffusion. Characteristically, the current–voltage curves did not show a decrease in the current at electrode polarization rates lower than 0.1 V/s. With the use of potentiostatic electrolysis at wave potentials and new phases diagnostics, reaction schemes were proposed. Anodic process at the first wave potential consists in a decrease in the Me:S ratio in the sulfide due to the oxidation of ferrous iron with the formation of X-ray amorphous Fe(III) hydroxides and metastable sulfide Fe4.5-nNi4.5S8. At the second wave potential, the metastable sulfide phase dissolves with the formation of elemental sulfur and ferric hydroxides. The concentration of nickel ions in the solution after electrolysis at the second wave potential exceeds the concentration of iron ions by approximately an order of magnitude. At more positive potentials, sulfate ions form.
2.4. Lead Sulfide (Galena)
Electrochemical properties of galena are described in a number of papers, mainly in relation to flotation processes [5][6][63][64][65][66][67][68][69][70]. It is noted that during the anodic polarization of galena in the pH range of 6.8–11, a layer of lead and sulfur oxide is formed, which inhibits the process of further oxidation [5]. It is emphasized that, according to a number of studies, the main product of galena oxidation in alkaline solutions is thiosulfate. Formation of S2O32− ions was reported in [67][70]. Using FTIR spectroscopy, in situ surface compounds on galena at anodic polarization in the range from −0.5 to +0.7 V (relative to a SHE) in an oxygen-free solution of sodium tetraborate (pH 9.18) were identified in [64]. A conclusion was made about the formation of lead hydroxide on the surface of galena by the salt-film mechanism. At the next stage of oxidation lead thiosulfate and lead sulfite formed.
An electrochemical study of the surface of galena in solutions of various acids was carried out and it was shown that passivation is not associated with shielding of the electrode surface with sulfur, salts, and lead oxides, and that ligands are directly involved in electrochemical reactions [63].
A detailed study of the electrochemical behavior of galena in acidic media was undertaken by Mikhlin in [71]. The cyclic current–voltage curves of galena show that the shape of the anodic curves and the values of the oxidation currents are fundamentally different in different acids, and that in solutions of sulfuric and hydrochloric, perchloric and nitric acids, the values of the maximum anodic oxidation currents are pairwise comparable. A feature of the anodic curves, common for sulfate and perchlorate solutions, in contrast to chloride and nitric acid solutions, is a significantly lower oxidation rate at low potentials (less than 0.5 V). Unusually, in sulfuric acid, the maximum at a potential of 0.7–0.8 V is reproduced in the reverse section of the curve. The electrochemical behavior of PbS is affected even by small additions of ligands, which do not change the activity of other ions and the solubility of lead sulfate and its other salts. Specifically, a number of anions, including those that do not form stable complexes with lead, for example, perchlorate ions, first increase the anodic currents, and then cause them to drop (passivation) and rise again with a further increase in the concentration of ClO4−. The cathode branches of the curves in different electrolytes are more similar and are characterized by a small maximum near 0.2 V, a more intense near −0.2 V, and an increase in current at potentials below −0.4 V. Cathode current is not directly related to an increase in the time and amount of anode oxidation products; longer oxidation leads to a decrease in the reduction charge. Specifically, the charge that passes through a stationary PbS electrode during cathodic potential sweep (up to −0.35 V) after anodic polarization, carried out in the potentiostatic mode in the passivation region, has two maxima corresponding to sulfuric and perchloric acids—at pre-oxidation times of the order of 10−2 s and ~100 s, while there is only one in hydrochloric acid electrolyte, at 10–50 s. The processes taking place over a time of about 10 ms are associated with the probable reaction of the transition of lead into solution and depletion of the surface of the sulfide phase in lead:
and charging of the resulting defects [71].
PbS → xPb2+ + Pb1xS + 2e−
2.5. Zinc Sulfide (Sphalerite)
Sphalerite is the most important zinc mineral, which contains various impurities. Common impurities in sphalerite are iron, lead, cadmium, and copper. Sphalerite is characterized by a high resistivity of 109–1012 Ω·cm [72]. To improve conductivity, carbon paste electrodes are often used in research. Impurities have a significant effect on the conductivity of a mineral.
A number of publications are devoted to the study of the electrochemical properties of sphalerite [72][73][74][75][76][77][78]. In particular, the anodic behavior of sphalerite is divided into two separate potential regions: dissolution in the passivation region and active dissolution. In the area of passivation, sphalerite is oxidized to zinc ions and elemental sulfur, which forms a passive layer on the surface of the mineral. In the active region, sphalerite dissolves without the formation of any passive layer.
In a later paper [79], dissolution of a sphalerite concentrate in a 0.5 M solution of sulfuric acid was studied using a carbon paste electrode at 22 °C. Various electrochemical methods were used, including cyclic voltammetry, chronoamperometry, and electrochemical impedance spectroscopy. In general, the study refines and updates the results obtained earlier. It was found that the oxidation of sphalerite proceeds in the range of anodic potentials up to 900 mV relative to the Ag/AgCl reference electrode. At low potentials (up to 400 mV), a sulfur-rich passive layer forms on the surface of the mineral. The predominant reaction on the electrode surface at 400 mV is the oxidative dissolution of sphalerite, in which zinc ions form as a result of an electrochemical reaction and a layer of elemental sulfur as a passive layer. As the potential increases to 550 mV, the passive layer partially dissolves. A further increase in the potential to 900 mV leads to the complete dissolution of the passive film and active dissolution of sphalerite.
Electrochemical behavior of natural polycrystalline sphalerite from the Zhairem deposit, which contained up to 4% iron in acid media and a borate buffer solution, was investigated in [71]. It was found that preliminary repeated acid etching of compact electrodes increases their conductivity and makes electrochemical experiments possible. The features of the current–voltage curves of sphalerite are a rapid increase in current with a cathodic potential shift and a slower one, often with a pronounced limiting current, with anodic polarization, which is typical for n-type semiconductors. In acid solutions, in some cases, there is a close to linear relationship between the anodic current and potential.
Thus, based on the analysis of studies of the processes of electrochemical oxidation of minerals, it can be concluded that the main products of oxidation on the surface of sulfides are elemental sulfur, metal oxides, and hydroxides, while metal ions and various oxidized sulfur compounds, mainly thiosulfate and sulfate ions, pass into the aqueous solution.
References
- Plaksin, I.N.; Shafeev, R.S.; Chanturiya, V.A. The relationship between the energy structure of mineral crystals and their flotation properties. In Proceedings of the VIII International Mineral Processing Congress; Mekhanobr: Leningrad, Russia, 1969; Volume 2, pp. 235–245. (In Russian).
- Plaksin, I.N.; Shafeev, R.S. Effects of some semiconductor properties of the surface on the interaction of xanthate with galena. Dokl. Akad. Nauk. USSR 1960, 132, 399–401. (In Russian)
- Plaksin, I.N.; Shafeev, R.S. On the effects of the electrochemical potential on the distribution of xanthate across the surface of sulfides. Dokl. Akad. Nauk. USSR 1958, 118, 546–548. (In Russian)
- Abramov, A.A.; Avdohin, V.M. Oxidation of Sulfide Minerals in Benefication Processes; Gordon and Breach Science Publishers: Philadelphia, PA, USA, 1997; 321p.
- Chanturiya, V.A.; Vigdergauz, V.E. Electrochemistry of Sulfides. Theory and Practice of Flotation; Publishing House Ore and Metals: Moscow, Russia, 2009; 280p.
- Buckley, A.N.; Hamilton, I.C.; Woods, R. Flotation of sulphide minerals. In Developments in Mineral Processing; Forssberg, K.S.E., Ed.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 41–115.
- Woods, R. Flotation of sulfide minerals. In Reagents in Mineral Technology; Somasundaran, P., Moudgil, B.M., Eds.; Marcel Dekker: New York, NY, USA, 1987; pp. 39–78.
- Bozkurt, V.; Xu, Z.; Finch, J.A. Pentlandite/pyrrhotite interaction and xanthate adsorbtion. Int. J. Miner. Process. 1998, 52, 203–214.
- Chanturiya, V.A. Scientific foundations of the electrochemical technology of mineral processing processes. Vestn. Akad. Nauk. USSR 1985, 9, 39–47. (In Russian)
- Chanturiya, V.A.; Nazarova, G.N. Electrochemical Technology for Beneficiation and Hydrometallurgical Processes; Nauka: Moscow, Russia, 1977; 160p. (In Russian)
- Watling, H.R. Review of biohydrometallurgical metals extraction from polymetallic mineral resources. Minerals 2015, 5, 1–60.
- Muñoz, A.; Bevilaqua, D.; Garcia, O., Jr. Leaching of Ni and Cu from mine wastes (tailings and slags) using acid solutions and A. Ferrooxidans. Adv. Mater. Res. 2009, 71–73, 425–428.
- Riekkola-Vanhanen, M.; Palmu, L. Talvivaara Nickel Mine–from a project to a mine and beyond. In Proceedings of the Symposium Ni-Co 2013, San Antonio, TA, USA, 3–7 March 2013; pp. 269–278.
- Tanne, C.K.; Schippers, A. Electrochemical applications in metal bioleaching. Adv. Biochem. Eng. Biotechnol. 2017, 167, 327–359.
- Tanne, C.K.; Schippers, A. Electrochemical process engineering in biohydrometallurgical metal recovery from mineral sulfides. Solid State Phenom. 2017, 262, 118–121.
- Holmes, P.R.; Crundwell, F.K. Kinetic aspects of galvanic interactions between minerals during dissolution. Hydrometallurgy 1995, 39, 353–375.
- Kalinnikov, V.T.; Makarov, D.V.; Makarov, V.N. Oxidation sequence of sulfide minerals in operating and out-of-service mine waste storage. Theor. Found. Chem. Eng. 2001, 35, 63–68.
- Parbhakar-Fox, A.; Lottermoser, B.; Bradshaw, D. Evaluating waste rock mineralogy and microtexture during kinetic testing for improved acid rock drainage prediction. Miner. Eng. 2013, 52, 111–124.
- Chopard, A.; Plante, B.; Benzaazoua, M.; Bouzahzah, H.; Marion, P. Geochemical investigation of the galvanic effects during oxidation of pyrite and base-metals sulfides. Chemosphere 2017, 166, 281–291.
- Kwong, Y.T.J.; Swerhone, G.W.; Lawrence, J.R. Galvanic sulphide oxidation as a metal-leaching mechanism and its environmental implications. Geochem. Explor. Environ. Anal. 2003, 3, 337–343.
- Radyushkina, K.A.; Wigdergauz, V.E.; Tarasevich, M.R.; Chanturiya, V.A. Electrochemistry of sulfide minerals. Electrochemical processes on the surface of pyrite and pyrrhotite in aqueous solutions of electrolytes. Elektrokhimjya 1986, 22, 1394–1398. (In Russian)
- Chanturiya, V.A.; Makarov, V.N.; Makarov, D.V.; Vasil’eva, T.N.; Belyaevskii, A.T. Electrochemical oxidation of pyrrhotine in alkaline media. Russ. J. Electrochem. 1999, 35, 759–763.
- Andriamanana, A.; Lamache, M. Etude electrochimique de la pyrite en milieu acide. Electrochim. Acta 1983, 28, 177–183.
- Hamilton, I.C.; Woods, R. An investigation of surface oxidation of pyrite and pyrrhotite by linear potential sweep voltammetry. J. Electroanal. Chem. 1979, 101, 327–343.
- Mischra, K.K.; Osseo-Asare, K. Aspects of the interfacial electrochemistry of semiconductor pyrite (FeS2). J. Electrochem. Soc. 1988, 135, 2502–2509.
- Moses, C.O.; Nordström, D.K.; Herman, J.S.; Mills, A.L. Aqueous pyrite oxidation by dissolved oxygen and by ferric ion. Geochim. Cosmochim. Acta 1987, 51, 1561–1571.
- Yin, Q.; Kelsall, G.H.; Vaughan, D.J.; Welham, N.J. Rotating ring(Pt)-disc(FeS2) electrode behavior in hydrocloric solutions. J. Colloid Interface Sci. 1999, 210, 375–383.
- Kelsall, G.H.; Yin, Q.; Vaughan, D.J.; England, K.E.R.; Brandon, N.P. Electrochemical oxidation of pyrite (FeS2) in aqueous electrolytes. J. Electroanal. Chem. 1999, 47, 116–125.
- Wang, X.-H. Interfacial electrochemistry of pyrite oxidation and flotation. J. Colloid Interface Sci. 1996, 178, 628–637.
- Chernyshova, I.V. Pyrite oxidation mechanism in aqueous solutions: An in situ FTIR study. Russ. J. Electrochem. 2004, 40, 69–77.
- Holmes, P.R.; Crundwell, F.K. Polysulfides do not cause passivation: Results from the dissolution of pyrite and implications for other sulfide minerals. Hydrometallurgy 2013, 139, 101–110.
- Mikhlin, Y. Reactivity of pyrrhotite surfaces: An electrochemical study. Phys. Chem. Chem. Phys. 2000, 2, 5672–5677.
- Kuklinskii, A.V.; Mikhlin, Y.L.; Pashkov, G.L.; Kargin, V.F.; Asanov, I.P. Conditions for the formation of a nonequilibrium nonstoichiometric layer on pyrrhotite in acid solutions. Russ. J. Electrochem. 2001, 37, 1269–1276.
- Mikhlin, Y.L.; Kuklinskii, A.V.; Pashkov, G.L.; Asanov, I.P. Pyrrhotite electrooxidation in acid solutions. Russ. J. Electrochem. 2001, 37, 1277–1282.
- Habashl, F.; Torres-Âcuna, N. The anodic dissolution of copper(l) sulfide and the direct recovery of copper and elemental sulfur from white metal. Trans. Metall. Eng. AIME 1968, 242, 780–787.
- Nechvoglod, O.V.; Pikalov, S.M. Mechanism of passive layer formation during electrochemical oxidation of copper(I) sulfide. Russ. J. Electrochem. 2021, 57, 580–586.
- O’Dell, C.S.; Walker, G.W.; Richardson, P.E. Electrochemistry of the chalcocite-xanthate system. J. Appl. Electrochem. 1996, 16, 544–554.
- Ghali, E.; Lewenstam, A. Electrodissolution of synthetic covellite in hydrochloric acid. J. Appl. Electrochem. 1982, 12, 369–376.
- Wigdergauz, V.E.; Chanturiya, V.A.; Teplyakova, M.V. Potentiometric study of chalcopyrite electroleaching. In Combined Ore Processing Methods; IPKON, Academy of Sciences of the USSR: Moscow, Russia, 1988; pp. 13–22. (In Russian)
- Biegler, T.; Swift, D.A. Anodic electrochemistry of chalcopyrite. J. Appl. Electrochem. 1979, 9, 545–554.
- Biegler, T.; Swift, D.A. The electrolytic reduction of chalcopyrite in acid solution. J. Appl. Electrochem. 1977, 6, 229–235.
- Radyushkina, K.A.; Wigdergauz, V.E.; Tarasevich, M.R.; Chanturiya, V.A. Electrochemistry of sulfide minerals. Surface redox transformations of chalcopyrite and chalcocite in aqueous solutions of electrolytes. Electrochimiya 1986, 22, 1491–1496. (In Russian)
- Chander, S. Electrochemistry of sulfide flotation: Growth characteristics of surface coating and their properties, with special reference to chalcopyrite and pyrite. Int. J. Miner. Process. 1991, 33, 121–134.
- Velásquez, P.; Gómez, H.; Ramos-Barrado, J.R.; Leinen, D. Voltammetry and XPS analysis of a chalcopyrite CuFeS2 electrode. Colloids Surf. A 1998, 140, 369–375.
- Velásquez, P.; Gómez, H.; Leinen, D.; Ramos-Barrado, J.R. Electrochemical impedance spectroscopy of chalcopyrite CuFeS2 electrode. Colloids Surf. A 1998, 140, 177–182.
- Moyo, T.; Petersen, J.; Nicol, M.J. The electrochemistry and kinetics of the oxidative dissolution of chalcopyrite in ammoniacal solutions. Part I–Anodic reactions. Hydrometallurgy 2018, 182, 97–103.
- Moyo, T.; Petersen, J.; Nicol, M.J. The electrochemistry and kinetics of the oxidative dissolution of chalcopyrite in ammoniacal solutions. Part II–Cathodic reactions. Hydrometallurgy 2019, 184, 67–74.
- Nicol, M.J. The electrochemistry of chalcopyrite in alkaline solutions. Hydrometallurgy 2019, 187, 134–140.
- Price, D.C.; Chilton, J.P. The anodic reactions of bornite in sulfuric acid solution. Hydrometallurgy 1981, 7, 117–133.
- Zhao, H.; Huang, X.; Hu, M.; Zhang, C.; Zhang, Y.; Wang, J.; Qin, W.; Qiu, G. Insights into the surface transformation and electrochemical dissolution process of bornite in bioleaching. Minerals 2018, 8, 173.
- Ásbjornsson, J.; Kelsall, G.H.; Pattrick, R.A.D.; Vaughan, D.J.; Wincott, P.L.; Hope, G.A. Electrochemical and surface analytical studies of enargite in acid solution. J. Electrochem. Soc. 2004, 151, E250–E256.
- Ma, Y.; Yang, Y.; Skinner, W.; Chen, M. Electrochemical and spectroscopic analysis of enargite (Cu3AsS4) dissolution mechanism in sulfuric acid solution. Hydrometallurgy 2020, 194, 105346.
- Nicol, M.J.; Ruiz-Sanchez, A.; Senanayake, G.; Tjandrawan, V.; Lapidus, G.T. The electrochemical behaviour of enargite in ammoniacal solutions. I. Anodic reactions. Hydrometallurgy 2019, 188, 92–100.
- Nicol, M.J.; Ruiz-Sanchez, A.; Senanayake, G.; Tjandrawan, V.; Lapidus, G.T. The electrochemical behaviour of enargite in ammoniacal solutions. II. Cathodic reactions. Hydrometallurgy 2019, 189, 105115.
- Buckley, A.N.; Woods, R. Electrochemical and XPS studies of the surface oxidation of synthetic heazlewoodite (Ni3S2). J. Appl. Electrochem. 1991, 21, 575–582.
- Power, G.P. The electrochemistry of the nickel sulfides. 1. NiS. Aust. J. Chem. 1981, 34, 2287–2291.
- Power, G.P. The electrochemistry of the nickel sulfides–2. Ni3S2. Electrochim. Acta 1982, 27, 359–364.
- Aromaa, J. Electrochemical dissolution of synthetic heazlewoodite (Ni3S2). Physicochem. Probl. Miner. Processing 2011, 46, 51–64.
- Thornber, M.R. Mineralogical and electrochemical stability of the nickel –iron sulphides–pentlandite and violarite. J. Appl. Electrochem. 1983, 13, 253–267.
- Warner, T.E.; Rice, N.M.; Taylor, N. An electrochemical study of the oxidative dissolution of synthetic pentlandite in aqueous media. Hydrometallurgy 1992, 31, 55–90.
- Warner, T.E.; Rice, N.M.; Taylor, N. Electrochemical study of oxidative dissolution of synthetic violarite in aqueous media. In Hydrometallurgy’94; Springer: Berlin/Heidelberg, Germany, 1994; pp. 273–287.
- Makarov, D.V.; Forsling, W.; Makarov, V.N. Electrooxidation of pentlandite in a carbonate solution. Russ. J. Electrochem. 2004, 40, 420–423. (In Russian)
- Mikhlin, Y.L.; Galkin, P.S.; Kopteva, S.A. Electrochemical study of galena surfaces in acid solutions. Izv. Sib. Otd. Akad. Nauk. SSSR. Ser. Him. 1988, 2, 11–17. (In Russian)
- Chernyshova, I.V. In situ study of the oxidation of galenite (natural PbS) in alkaline media by FTIR spectroscopy: Anode processes in the absence of oxygen. Russ. J. Electrochem. 2001, 37, 579–584.
- Ahlberg, E.; Elfstrom, B.A. Anodic polarization of galena in relation to flotation. Int. J. Miner. Process. 1997, 33, 135–142.
- Cisneros-Gonzalez, I.; Oropeza-Guzman, M.T.; Gonzalez, I. Cyclic voltammetry applied to the characterisation of galena. Hydrometallurgy 1999, 53, 133–144.
- Gardner, J.R.; Woods, R. Study of surface oxidation of galena using cyclic voltammetry. J. Electroanal. Chem. 1979, 100, 447–459.
- Nicol, M.J.; Paul, R.L.; Diggle, J.W.; Saunders, A.P. The electrochemical behaviour of galena (lead sulphide): II. Catodic reduction. Electrochim. Acta 1978, 23, 635–639.
- Paul, R.L.; Nicol, M.J.; Diggle, J.W.; Saunders, A.P. The electrochemical behaviour of galena (lead sulphide): I. Anodic dissolution. Electrochim. Acta 1978, 23, 625–633.
- Pritzker, M.D.; Yoon, R.H. Thermodynamic calculation on sulfide flotation system. 2. Comparison with electrochemical experiments on the galena-ethylxanthate system. Int. J. Miner. Process. 1987, 20, 267–290.
- Mikhlin, Y.L. State of the Real Surface and Features of the Kinetics of Dissolution and Oxidation of Metal Sulfides in Interaction with Acid Solutions. Extended abstract. Ph.D. Thesis, Institute of Chemistry and Chemical Technologies of the SB RAS, Krasnoyarsk, Russia, 2003; 40p. (In Russian).
- Ahlberg, E.; Ásbjörnsson, J. Carbon paste electrodes in mineral processing: An electrochemical study of sphalerite. Hydrometallurgy 1994, 36, 19–37.
- Choi, W.K.; Torma, A.E.; Ohline, R.W.; Ghali, E. Electrochemical aspects of zinc sulphide leaching by Thiobacillus ferrooxidans. Hydrometallurgy 1993, 33, 137–152.
- Lotens, J.P.; Wesker, E. The behaviour of sulphur in the oxidative leaching of sulphidic minerals. Hydrometallurgy 1987, 18, 39–54.
- Nava, J.L.; Oropeza, M.T.; Gonzalez, I. Oxidation of Mineral Species as a Function of the Anodic Potential of Zinc Concentrate in Sulfuric Acid. J. Electrochem. Soc. 2004, 151, B387–B393.
- Narasagoudar, R.A.; Johnson, J.W.; O’Keefe, T.J. The anodic dissolution of ZnS electrodes in sulfuric acid solutions. Hydrometallurgy 1982, 9, 37–55.
- Srinivasan, G.N.; Iyer, S.V. Cyclic voltammetric studies on sphalerite electrodes. Bull. Electrochem. 2000, 16, 5–9.
- Urbano, G.; Meléndez, A.M.; Reyes, V.E.; Veloz, M.A.; González, I. Galvanic interactions between galena–sphalerite and their reactivity. Int. J. Miner. Process. 2007, 82, 148–155.
- Karimia, S.; Ghahreman, A.; Rashchia, F.; Moghaddam, J. The mechanism of electrochemical dissolution of sphalerite in sulfuric acid media. Electrochim. Acta 2017, 253, 47–58.
More