In eukaryotes, genomic DNA is bound with histone proteins and packaged into chromatin. The nucleosome, a fundamental unit of chromatin, regulates the accessibility of DNA to enzymes involved in gene regulation. SDuring the past few years, structural analyses of chromatin architectures have been limited to evolutionarily related organisms. The amino acid sequences of histone proteins are highly conserved from humans to yeasts, but are divergent in the deeply branching protozoan groups, including human parasites that are directly related to human health. Certain large DNA viruses, as well as archaeal organisms, contain distant homologs of eukaryotic histone proteins. The divergent sequences give rise to unique and distinct nucleosome architectures, although the fundamental principles of histone folding and DNA contact are highly conserved.
- nucleosome
- chromatin
- parasite
- DNA virus
1. Introduction
2. Structures of Histone Complexes in Nucleosomes
2.1. Histone Fold
Nucleosome formation is similar from humans to yeast, and even to Archaea, as the basic structures of the folded histones are shared in various species. In the nucleosome, the central ‘disc’ architecture is composed of four core histones, H2A, H2B, H3, and H4, sharing well-conserved motifs termed histone-fold domains comprising three α-helices, α1, α2, and α3, separated by two loops, L1 and L2 [22][23] (Figure 1A). The H2A–H2B and H3–H4 heterodimers are assembled into the characteristic handshake motif via central hydrophobic interactions, along with electrostatic contacts and hydrogen bonds [8] (Figure 1B,C). Two H3–H4 dimers are joined into a (H3–H4)2 tetramer in physiological ionic strength solutions by the interactions of the C-terminal halves of the α2 helices of H3 and H3′ (adjacent copy of H3) and the α3 helix of H3. A single H3–H4 tetramer and two H2A–H2B dimers are incorporated into the nucleosome core particle to form the histone octamer, around which the double-stranded DNA is wrapped (Figure 1D). Within the octamer, the H2A–H2B dimers are assembled by the interaction of H2B with H4, in addition to the H3–H3′ interaction. These remarkable associations are described as the H2B–H4 four-helix bundle and the H3–H3′ four-helix bundle, respectively [8].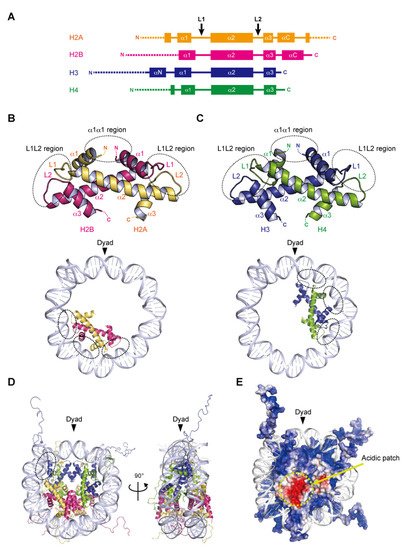
2.2. DNA Wrapping around the Histone Octamer
In the nucleosome, 145–147 base pairs of DNA are wrapped around the histone octamer in 1.65 left-handed superhelical turns with 14 contact sites on the histone surface, where the DNA phosphate backbone faces the histone octamer. Twelve of the 14 DNA–histone contact sites are mediated by the handshake motifs, which are divided into two types: the α1α1 and L1L2 regions that are enriched in positively charged residues (Figure 1B,C). The α1α1 region is formed by the N-terminal ends of the α1 helices of the histone heterodimer pair (Figure 1B,C). The L1L2 region is formed by the L1 and L2 loops and the C-termini of the α2 helices on each end of the heterodimer pair (Figure 1B,C). A total of four α1α1 and eight L1L2 regions interact directly with 12 helical turns (120 bp) of the DNA in the minor grooves. The two remaining DNA–histone contact sites originate from the N-terminal α helix (αN) and the N-terminal extension of H3, where the positively charged residues bind to the DNA at the entry–exit sites of the nucleosome (Figure 1D). Related to the studies of histone variants, some mutational analyses have clearly shown the contributions of arginine residues, at positions 42 [24], 49, and 53 [25] on the αN and N-terminal extension of H3, to the DNA flexibility at the entry–exit sites. Additionally, the interaction of the C-terminal region of H2A with the H3–H4 tetramer guides this last turn of the nucleosomal DNA (Figure 1D). The histone variant H2A.B (formerly H2A.bbd) lacking the C-terminal region forms a nucleosome with ~110 base pairs of DNA wrapped around the octamer [26][27]. The flexibility of the DNA at the entry–exit sites also plays a considerable role in the higher-order structure of chromatin.2.3. Acidic Patch of the Nucleosome
In many eukaryotic nucleosomes, the acidic patch is a fundamental and well-conserved region located on the disc surface. The acidic side chains of amino acid residues in the α2 helix of H2A (Glu56, Glu61, Glu64, Asp90, Glu91, and Glu92 in human) and the αC helix of H2B (Glu105 and Glu113 in human) form a negatively charged patch exposed to the solvent (Figure 1E). The acidic patch acts as a binding platform for many proteins that regulate chromatin function via arginine anchoring, including histone modification enzymes, chromatin remodelers, and DNA methyltransferases [6][28]. In addition, X-ray crystallographic analyses have shown that the basic residues in the N-terminal tail of H4 contact the acidic patch, in a manner reminiscent of the nucleosome–nucleosome interactions in nucleosome arrays, suggesting that the acidic patch may also be involved in chromatin compaction [28][29].2.4. Histone Tails
Histone proteins contain intrinsically disordered regions at the N-termini of H2A, H2B, H3, and H4, and the C-terminus of H2A (Figure 1A,E). These flexible ‘tails’, which protrude from the central disc architecture of the nucleosome core, are partly exposed to the solvent. With their many positively charged residues, such as lysine and arginine, the histone tails associate with the negatively charged DNA in the nucleosome core particle or linker regions [30][31]. However, the histone tails do not seem to affect the disc architecture of the nucleosome, as suggested from the X-ray structures of N-terminally truncated histones [32]. In contrast, the histone tails likely alter the higher-order structure of chromatin through the interactions between the H4 tail and the acidic patch [33]. Biophysical evidence has indicated the contribution of histone tails to the compaction of nucleosome arrays [34]. Importantly, the histone tails are often enzymatically modified in vivo, by the acetylation, methylation, and ubiquitylation of lysine residues, and the phosphorylation of serine and threonine residues [35]. These post-translational modifications serve as binding sites for factors that regulate the chromatin structure and function [5][36][37]. Although the histone tail sequences are relatively diverse among eukaryotic species, the residues subjected to post-transcriptional modifications are highly conserved. In particular, the H3 Lys4, Lys9, Lys27, Lys36, and H4 Lys20 residues, which are targeted for methylation and acetylation, are inferred to be conserved lysine residues even in the last eukaryotic common ancestor (LECA) [38].3. Structures of Nucleosomes Containing Parasite Histones
3.1. Genomic Organization and Gene Regulation in Giardia
Giardia lamblia is a flagellated unicellular eukaryote that parasitizes the small intestines of humans and animals. Giardia causes a diarrheal disease termed giardiasis, which is one of the most common parasitic infections throughout the world [39][40]. The Giardia life cycle consists of two main forms: the non-infectious trophozoite that causes the primary symptoms of diarrhea by proliferating on the surfaces of the host intestinal cells, and the highly infective cyst form, which is environmentally resistant and can survive outside the host. This parasite is classified in the order diplomonadia within the Excavata supergroup. In the eukaryotic tree of life, Giardia is in a deeply branched position, with a simplification of most cellular processes, and lacking certain organelles, such as mitochondria and peroxisomes [41]. Giardia has two nuclei with equivalent activities. Consistent with higher eukaryotes, genomic regulation occurs on the “beads-on-a-string” chromatin structure [42]. The 12 mega base pairs of the Giardia genome contain 4963 open reading flames with few introns, short intergenic and 3′ and 5′ untranslated regions, and small promoter regions that are close to transcriptional start sites [43][44][45]. Giardia has a simple transcriptional initiation complex [43], with few transcriptional factors [46] and chromatin remodeler subunits [47]. A genome-wide transcriptional analysis indicated that bidirectional transcription produces both sense and antisense transcripts in Giardia [48]. In addition, Giardia possesses an RNA interference system, which is involved in antigenic switching, conferring a pathogenic ability to evade the host immune system [49]. These results suggest that transcriptional control is limited in Giardia. In the encystation process, the transformation from trophozoite to cyst stage, histone modifications such as ubiquitylation, deacetylation, and methylation have been verified, indicating that the chromatin-based regulation of gene expression is essential for biological processes in Giardia [50][51][52][53][54].3.2. Structure of the Nucleosome Containing Giardia Histones
The Giardia genome encodes two copies of the H2A, H2B, and H3 genes, and three copies of the H4 genes for canonical histones, and their corresponding mRNAs are polyadenylated [44][55]. Histone variants H3B and CenH3 localized in the centromere were identified, but not the linker histone H1 [56]. In higher eukaryotes, the phosphorylation of an H2A variant, the H2A.X Ser139 residue (γH2A.X), is responsible for facilitating DNA repair in response to double-stranded breaks [15][36][57]. Although no H2A variant has been identified in Giardia, the “Ser-Gln-Asp-Leu” motif, within the H2A.X variant in higher eukaryotes, is present in the H2A C-terminus. The four core histones share the typical eukaryotic features of histone-fold domains, containing three α helices separated by two loops [55]. Parasite histones generally have diverse amino acid sequences, with remarkably low sequence identity compared to metazoan histones [58][59]. Indeed, the identities of the Giardia lamblia (G. lamblia) histone-fold domains of H2A, H2B, H3, and H4 compared to the human histone-fold domains are 48, 49, 60, and 78%, respectively. As in other eukaryotes, the Giardia core histones contain intrinsically disordered “tail” regions that undergo conserved post-translational modifications. Emery-Corbin and colleagues identified Giardia histone modifications, including methylation, acetylation, and phosphorylation, using mass spectrometry [60]. They demonstrated that Giardia histone tails are highly modified (more than 50 sites were identified) and that these histone modifications in Giardia are largely equivalent to those in many other eukaryotes, suggesting that similar epigenetic mechanisms exist in this parasite [60]. Among the well-conserved modifications, the phosphorylation of H3 Ser10, and the methylations of H3 Lys4, Lys36, Lys9 and Lys27, but not H3 Lys79 and H4 Lys20, were identified in Giardia. The cryo-EM structure of the G. lamblia nucleosome was determined at a 3.6 angstrom resolution, after reconstitution with 145 base pairs of modified Widom 601 (601L) palindromic DNA [61]. The overall structure of the G. lamblia nucleosome resembles that of the human nucleosome (Figure 2A). All G. lamblia histones fold into the characteristic handshake motifs, but with notable deviations from the main chains of human histones in the nucleosome, including the G. lamblia-specific insertions of six and two residues in H2B and H3, respectively (Figure 2B,C). This six-residue insertion extends the α1 helix and L1 loop of G. lamblia H2B and alters the conformation of the L1L2 region (Figure 2B). Importantly, the insertion affects the shape and peptide-binding properties of the adjacent acidic patch (see below). In the cryo-EM structure of the G. lamblia nucleosome, only 125 base pairs of DNA are wrapped asymmetrically around the histones, and the DNA flexibility was confirmed in solution (Figure 2A). Furthermore, in the cryo-EM structure of the G. lamblia nucleosome, only one of the shortened αN helices of H3 was resolved, and the two C-terminal regions of H2A were not visualized (Figure 2D,E). In the human canonical nucleosome, the αN helix and N-terminal extension of H3 bind directly to the last turn of the superhelical DNA, and these interactions are considered to be guided by the C-terminal region of H2A via a hydrophobic cluster. In G. lamblia, the hydrophobic residues of the H2A C-terminal region are replaced by hydrophilic or small aliphatic residues. A mutational analysis revealed that the C-terminal region of G. lamblia H2A is responsible for the flexibility of the DNA entry–exit sites in the nucleosome. Consistent with this DNA flexibility, the nucleosome array containing G. lamblia histones appears to adopt a more relaxed conformation, compared to the human nucleosome array. In addition, a biological analysis revealed the instability of the G. lamblia nucleosome. These properties of relaxed chromatin and nucleosome instability could facilitate transcriptional activation in Giardia, which possesses simple gene regulatory systems.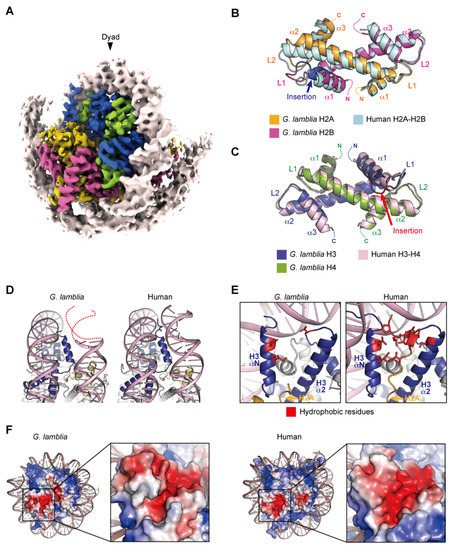
References
- Olins, A.L.; Olins, D.E. Spheroid chromatin units (ν bodies). Science 1974, 183, 330–332.
- Wolffe, A.P. Chromatin: Structure and Function, 3rd ed.; Academic Press: San Diego, CA, USA, 1998.
- Zlatanova, J.; Bishop, T.C.; Victor, J.M.; Jackson, V.; van Holde, K. The nucleosome family: Dynamic and growing. Structure 2009, 17, 160–171.
- Hargreaves, D.; Crabtree, G. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420.
- Musselman, C.A.; Lalonde, M.E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227.
- McGinty, R.K.; Tan, S. Principles of nucleosome recognition by chromatin factors and enzymes. Curr. Opin. Struct. Biol. 2021, 71, 16–26.
- Tsunaka, Y.; Kajimura, N.; Tate, S.; Morikawa, K. Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res. 2005, 33, 3424–3434.
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260.
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 2002, 319, 1097–1113.
- Harp, J.M.; Hanson, B.L.; Timm, D.E.; Bunick, G.L. Asymmetries in the nucleosome core particle at 2.5 Å resolution. Acta Crystallogr. D. Biol. Crystallogr. 2000, 56, 1513–1534.
- Clapier, C.R.; Chakravarthy, S.; Petosa, C.; Fernandez-Tornero, C.; Luger, K.; Muller, C.W. Structure of the Drosophila nucleosome core particle highlights evolutionary constraints on the H2A-H2B histone dimer. Proteins 2008, 71, 1–7.
- White, C.L.; Suto, R.K.; Luger, K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001, 20, 5207–5218.
- Fukushima, Y.; Hatazawa, S.; Hirai, S.; Kujirai, T.; Ehara, H.; Sekine, S.I.; Takizawa, Y.; Kurumizaka, H. Structural and biochemical analyses of the nucleosome containing Komagataella pastoris histones. J. Biochem. 2022, 172, 79–88.
- Suto, R.K.; Clarkson, M.J.; Tremethick, D.J.; Luger, K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000, 7, 1121–1124.
- Talbert, P.B.; Henikoff, S. Histone variants on the move: Substrates for chromatin dynamics. Nat. Rev. Mol. Cell. Biol. 2017, 18, 115–126.
- Kurumizaka, H.; Kujirai, T.; Takizawa, Y. Contributions of Histone Variants in Nucleosome Structure and Function. J. Mol. Biol. 2021, 433, 166678.
- Sandman, K.; Krzycki, J.A.; Dobrinski, B.; Lurz, R.; Reeve, J.N. HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc. Natl. Acad. Sci. USA 1990, 87, 5788–5791.
- Bailey, K.A.; Chow, C.S.; Reeve, J.N. Histone stoichiometry and DNA circularization in archaeal nucleosomes. Nucleic Acids Res. 1999, 27, 532–536.
- Mattiroli, F.; Bhattacharyya, S.; Dyer, P.N.; White, A.E.; Sandman, K.; Burkhart, B.W.; Byrne, K.R.; Lee, T.; Ahn, N.G.; Santangelo, T.J.; et al. Structure of histone-based chromatin in Archaea. Science 2017, 357, 609–612.
- Silverman, J.M.; Chan, S.K.; Robinson, D.P.; Dwyer, D.M.; Nandan, D.; Foster, L.J.; Reiner, N.E. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 2008, 9, R35.
- Bayer-Santos, E.; Aguilar-Bonavides, C.; Rodrigues, S.P.; Cordero, E.M.; Marques, A.F.; Varela-Ramirez, A.; Choi, H.; Yoshida, N.; Silveira, J.F.D.; Almeida, I.C. Proteomic analysis of Trypanosoma cruzi secretome: Characterization of two populations of extracellular vesicles and soluble proteins. J. Proteome Res. 2013, 12, 883–897.
- Finch, J.T.; Lutter, L.C.; Rhodes, D.; Brown, R.S.; Rushton, B.; Levitt, M.; Klug, A. Structure of nucleosome core particles of chromatin. Nature 1977, 269, 29–36.
- Arents, G.; Burlingame, R.W.; Wang, B.C.; Love, W.E.; Moudrianakis, E.N. The nucleosomal core histone octamer at 3.1 Å resolution: A tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA 1991, 88, 10148–10152.
- Ueda, J.; Harada, A.; Urahama, T.; Machida, S.; Maehara, K.; Hada, M.; Makino, Y.; Nogami, J.; Horikoshi, N.; Osakabe, A.; et al. Testis-specific histone variant H3t gene is essential for entry into spermatogenesis. Cell Rep. 2017, 18, 593–600.
- Kono, H.; Shirayama, K.; Arimura, Y.; Tachiwana, H.; Kurumizaka, H. Two arginine residues suppress the flexibility of nucleosomal DNA in the canonical nucleosome core. PLoS ONE 2015, 10, e0120635.
- Arimura, Y.; Kimura, H.; Oda, T.; Sato, K.; Osakabe, A.; Tachiwana, H.; Sato, Y.; Kinugasa, Y.; Ikura, T.; Sugiyama, M.; et al. Structural basis of a nucleosome containing histone H2A.B/H2A.Bbd that transiently associates with reorganized chromatin. Sci. Rep. 2013, 3, 3510.
- Zhou, M.; Dai, L.; Li, C.; Shi, L.; Huang, Y.; Guo, Z.; Wu, F.; Zhu, P.; Zhou, Z. Structural basis of nucleosome dynamics modulation by histone variants H2A.B and H2A.Z.2.2. EMBO J. 2021, 40, e105907.
- Kalashnikova, A.A.; Porter-Goff, M.E.; Muthurajan, U.M.; Luger, K.; Hansen, J.C. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J. R. Soc. Interface 2013, 10, 20121022.
- Chodaparambil, J.V.; Barbera, A.J.; Lu, X.; Kaye, K.M.; Hansen, J.C.; Luger, K. A charged and contoured surface on the nucleosome regulates chromatin compaction. Nat. Struct. Mol. Biol. 2007, 14, 1105–1107.
- Stützer, A.; Liokatis, S.; Kiesel, A.; Schwarzer, D.; Sprangers, R.; Söding, J.; Selenko, P.; Fischle, W. Modulations of DNA Contacts by Linker Histones and Post-translational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol. Cell 2016, 61, 247–259.
- Furukawa, A.; Wakamori, M.; Arimura, Y.; Ohtomo, H.; Tsunaka, Y.; Kurumizaka, H.; Umehara, T.; Nishimura, Y. Acetylated histone H4 tail enhances histone H3 tail acetylation by altering their mutual dynamics in the nucleosome. Proc. Natl. Acad. Sci. USA 2020, 117, 19661–19663.
- Iwasaki, W.; Miya, Y.; Horikoshi, N.; Osakabe, A.; Taguchi, H.; Tachiwana, H.; Shibata, T.; Kagawa, W.; Kurumizaka, H. Contribution of histone N-terminal tails to the structure and stability of nucleosomes. FEBS Open Bio. 2013, 3, 363–369.
- Luger, K.; Richmond, T.J. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 1998, 8, 140–146.
- Dorigo, B.; Schalch, T.; Bystricky, K.; Richmond, T.J. Chromatin fiber folding: Requirement for the histone H4 N-terminal tail. J. Mol. Biol. 2003, 327, 85–96.
- Cavalieri, V. The expanding constellation of histone post-translational modifications in the epigenetic landscape. Genes 2021, 12, 1596.
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications-cause and conseuence of genome function. Nat. Rev. Genet 2022. online ahead of print.
- Chan, J.; Kumar, A.; Kono, H. RNAPII driven post-translational modifications of nucleosomal histones. Trends Genet. 2022. online ahead of print.
- Aravind, L.; Burroughs, A.M.; Zhang, D.; Iyer, L.M. Protein and DNA modifications: Evolutionary imprints of bacterial biochemical diversification and geochemistry on the provenance of eukaryotic epigenetics. Cold Spring Harb. Perspect. Biol. 2014, 6, a016063.
- Ankarklev, J.; Jerlström-Hultqvist, J.; Ringqvist, E.; Troell, K.; Svärd, S.G. Behind the smile: Cell biology and disease mechanisms of Giardia species. Nat. Rev. Microbiol. 2010, 8, 413–422.
- Einarsson, E.; Ma’ayeh, S.; Svärd, S.G. An up-date on Giardia and giardiasis. Curr. Opin. Microbiol. 2016, 34, 47–52.
- Adam, R.D. Biology of Giardia lamblia. Clin. Microbiol. Rev. 2001, 14, 447–475.
- Tůmová, P.; Uzlíková, M.; Wanner, G.; Nohýnková, E. Structural organization of very small chromosomes: Study on a single-celled evolutionary distant eukaryote Giardia intestinalis. Chromosoma 2015, 124, 81–94.
- Morrison, H.G.; McArthur, A.G.; Gillin, F.D.; Aley, S.B.; Adam, R.D.; Olsen, G.J.; Best, A.A.; Cande, W.Z.; Chen, F.; Cipriano, M.J.; et al. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 2007, 317, 1921–1926.
- Yee, J.; Tang, A.; Lau, W.L.; Ritter, H.; Delport, D.; Page, M.; Adam, R.D.; Müller, M.; Wu, G. Core histone genes of Giardia intestinalis: Genomic organization, promoter structure, and expression. BMC Mol. Biol. 2007, 10, 8–26.
- Xu, F.; Jex, A.; Svärd, S.G. A chromosome-scale reference genome for Giardia intestinalis WB. Sci. Data 2020, 7, 38.
- Iyer, L.M.; Anantharaman, V.; Wolf, M.Y.; Aravind, L. Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int. J. Parasitol. 2008, 38, 1–31.
- Flaus, A.; Martin, D.M.A.; Barton, G.J.; Owen-Hughes, T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006, 34, 2887–2905.
- Teodorovic, S.; Walls, C.D.; Elmendorf, H.G. Bidirectional transcription is an inherent feature of Giardia lamblia promoters and contributes to an abundance of sterile antisense transcripts throughout the genome. Nucleic Acids Res. 2007, 35, 2544–2553.
- Prucca, C.G.; Slavin, I.; Quiroga, R.; Elías, E.V.; Rivero, F.D.; Saura, A.; Carranza, P.G.; Luján, H.D. Antigenic variation in Giardia lamblia is regulated by RNA interference. Nature 2008, 456, 750–754.
- Sonda, S.; Morf, L.; Bottova, I.; Baetschmann, H.; Rehrauer, H.; Caflisch, A.; Hakimi, M.A.; Hehl, A.B. Epigenetic mechanisms regulate stage differentiation in the minimized protozoan Giardia lamblia. Mol. Microbiol. 2010, 76, 48–67.
- Nino, C.A.; Chaparro, J.; Soffientini, P.; Polo, S.; Wasserman, M. Ubiquitination dynamics in the early-branching eukaryote Giardia intestinalis. Microbiologyopen 2013, 2, 525–539.
- Carranza, P.G.; Gargantini, P.R.; Prucca, C.G.; Torri, A.; Saura, A.; Svard, S.; Lujan, H.D. Specific histone modifications play critical roles in the control of encystation and antigenic variation in the early-branching eukaryote Giardia lamblia. Int. J. Biochem. Cell Biol. 2016, 81, 32–43.
- Salusso, A.; Zlocowski, N.; Mayol, G.F.; Zamponi, N.; Ropolo, A.S. Histone methyltransferase 1 regulates the encystation process in the parasite Giardia lamblia. FEBS J. 2017, 284, 2396–2409.
- Lagunas-Rangel, F.A.; Bermúdez-Cruz, R.M. Epigenetics in the early divergent eukaryotic Giardia duodenalis: An update. Biochimie 2019, 156, 123–128.
- Wu, G.; McArthur, A.G.; Fiser, A.; Sali, A.; Sogin, M.L.; Mllerm, M. Core histones of the amitochondriate protist, Giardia lamblia. Mol. Biol. Evol. 2000, 17, 1156–1163.
- Dawson, S.C.; Sagolla, M.S.; Cande, W.Z. The cenH3 histone variant defines centromeres in Giardia intestinalis. Chromosoma 2007, 116, 175–184.
- Downs, J.A.; Lowndes, N.F.; Jackson, S.P. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000, 408, 1001–1004.
- Malik, S.H.; Henikoff, S. Phylogenomics of the nucleosome. Nat. Struct. Biol. 2003, 10, 882–891.
- Patwal, I.; Trinh, H.; Golden, A.; Flaus, A. Histone sequence variation in divergent eukaryotes facilitates diversity in chromatin packaging. BioRxiv 2021.
- Emery-Corbin, S.J.; Hamey, J.J.; Balan, B.; Rojas-López, L.; Svärd, S.G.; Jex, A.R. Eukaryote-conserved histone post-translational modification landscape in Giardia duodenalis revealed by mass spectrometry. Int. J. Parasitol. 2021, 51, 225–239.
- Sato, S.; Takizawa, Y.; Hoshikawa, F.; Dacher, M.; Tanaka, H.; Tachiwana, H.; Kujirai, T.; Iikura, Y.; Ho, C.-H.; Adachi, N.; et al. Cryo-EM structure of the nucleosome core particle containing Giardia lamblia histones. Nucleic Acids Res. 2021, 49, 8934–8946.