Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Dale D. Tang and Version 3 by Beatrix Zheng.
Although asthma is classified into Th2-high and Th2-low asthma, the disease can be induced by mixed airway inflammation. Patients may have Th2-high asthma in the early stage and have Th2-low asthma in a later stage or vice versa; or Th2-high asthma and Th2-low asthma occur concurrently. Because of the complexity of asthma, we discuss the mechanisms of Th2-high asthma, Th2-low asthma, and other mechanisms separately.
- asthma
- smooth muscle
- cytokine
- inflammation
- biomarker
1. Mechanisms of Th2-High Asthma
Th2 cells are a distinct lineage of CD4+ effector T cells that secrete interleukin (IL)-4, IL-5, IL-13, and IL-9. Approximately 50% of mild-to-moderate asthma and a large portion of severe asthma is induced by Th2-dependent inflammation [1][2][1,2]. Since Th2-high asthma has been reviewed in detail elsewhere [2][3][4][2,3,4], thwe researchers ssummarize the key points for the mechanisms of Th-2 high asthma.
Th2 inflammation has two major phases: 1. Sensitization: When allergens enter the low airways, antigen-presenting cells process and present the allergens to Th2 cells, which secret Th2 cytokines, including IL-5, IL-4, and IL-13. IL-4 and IL-13 activate B cells, which produce IgE and bind to FcεRI of mast cells. 2. Challenge: When the same allergens enter the airways, they bind to IgE, which induces mast cells to release mediators, such as leukotrienes (LTs), histamine, and ILs. In addition, allergens act on cholinergic nerves to release acetylcholine. These mediators and neurotransmitters irritate airway smooth muscle and induce bronchoconstriction [1][2][3][1,2,3]. In addition, IL-5 facilitates eosinophil production, maturation, and recruitment to the lungs [5]. Eosinophils also release mediators, including major basic protein (MBP), which stimulates mast cells to release histamines and LTs. MBP also inhibits M2 receptor and promotes acetylcholine release from cholinergic nerves and induces bronchospasm [6]. Furthermore, IL-13 directly sensitizes airway smooth muscle contraction, stimulates epithelial cells to secret mucins, and induces fibrosis [7] (Figure 1).
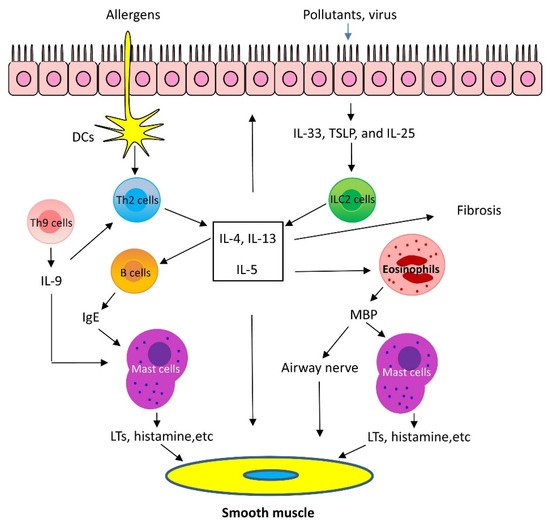
Figure 1. Mechanism of Th2-high asthma. When allergens enter the low airways, dendritic cells (DCs) present the allergens to Th2 cells, which secrete Th2 cytokines, including interleukin (IL)-5, IL-4, and IL-13. IL-4 and IL-13 activate B cells, which produce IgE. IgE subsequently binds to surface of mast cells. When the same allergens enter the airways, they interact with IgE, which induces mast cells to release mediators, such as leukotrienes (LTs), histamine, and ILs. These mediators irritate airway smooth muscle and induce bronchoconstriction. In addition, IL-5 facilitates eosinophil recruitment to the lungs. Eosinophils also release mediators, including major basic protein (MBP), which stimulates mast cells to release histamines and LTs. MBP also inhibits M2 receptor and promotes acetylcholine release from cholinergic nerves and induces bronchospasm. Furthermore, IL-13 directly sensitizes airway smooth muscle contraction, stimulates epithelial cells to secret mucins, and induces fibrosis. Th9 cells can secrete IL-9, which activates Th2 cells and promotes mast cell accumulation. Lastly, epithelium injury by infection and pollutants induces release of cytokines, including thymic stromal lymphopoietin (TSLP), IL-25, and IL-33, which activate type 2 innate lymphoid cells (ILC2) and produce Th2 cytokines, such as IL-5 and IL-13.
Recent studies demonstrated that the airway epithelium produces cytokines in response to injury, infection, and pollutants. These epithelial-derived cytokines include thymic stromal lymphopoietin (TSLP), IL-25, and IL-33. TSLP, IL-25, and IL-33 activate type 2 innate lymphoid cells (ILC2), which generate Th2 cytokines, such as IL-5 and IL-13 and induce Th2 lung inflammation [1][2][1,2]. Additionally, there is evidence to suggest that IL-33 may directly affect mast cell activation, airway smooth muscle migration, and asthma phenotype [8] (Figure 1).
Th9 cells and IL-9 are also involved in Th2 lung inflammation [9]. Th9 cells produce the cytokines IL-9, IL-10, and IL-21; however, IL-9 is likely to contribute to asthma pathology. Because of its pleiotropic effects, IL-9 influences a variety of distinct cell types, such as T cells, B cells, mast cells, and macrophages. IL-9 may promote Th2 inflammation by activating Th2 cells and by increasing mast cell accumulation [9]. IL-9 may also activate Arg1+ interstitial macrophages, which secrete the chemokine CCL5. CCL5 then recruits eosinophils, T cells, and monocytes into the lungs to propagate type 2 inflammation [10] (Figure 1).
Natural killer T (NKT) cells are a distinct subset of lymphocytes that are abundant in the lungs as well as lymphoid organs. It was proposed that NKT cells secrete IL-4 and IL-13 or facilitate Th2 cells to increase production of IL-4 and IL-13 [11]. However, other studies do not support this notion [12][13][12,13].
Regulatory T cells (Tregs) are a specific CD4+ T cell population that act to suppress immune response, thereby maintaining homeostasis and self-tolerance. Tregs have been classified based on the expression of the transcription factor FOXP3. Tregs may inhibit asthma pathogenesis by suppressing the activation/functions of ILC2, mast cells, antigen-presenting cells, Th1/Th2/Th17 cells, eosinophils, neutrophils, and B cells [14].
One of the targets of Th2 cytokines is periostin, a matricellular protein that is a dynamically expressed non-structural protein present in the extracellular matrix. Periostin expression is upregulated by IL-4 and IL-13 in cultured bronchial epithelial cells and bronchial fibroblasts [15] and is one of the most differentially expressed bronchial epithelial genes between asthmatic patients and healthy control subjects [16]. The role of periostin in asthma is still under investigation. There are reports to suggest that periostin supports adhesion and migration of IL-5-stimulated human eosinophils and Th2 inflammation in asthma [17]. On the other hand, other studies suggest that periostin plays a protective role, rather than detrimental role in asthma. Periostin positively regulates TGF-β production, which promotes T-regulatory cell differentiation. Differentiated T cells inhibit airway inflammation and IgE production [18].
2. Mechanisms of Th2-Low Asthma
2.1. IL-17
IL-17 has been proposed to play an important role in Th2-low asthma [19][20][21][19,20,21]. Variants in the IL-17 pathway genes may be related to asthma pathology [22][23][22,23]. Higher levels of IL-17 are found in serum, sputum, and bronchoalveolar lavage fluid (BALF) of patients with asthma, which is associated with asthma severity [19][20][19,20]. There are several cell types secreting IL-17 cytokines. CD4+ Th17 cells are one of the major sources of IL-17. Other cellular sources include major histocompatibility complex class I-restricted CD8+ T-cells, Natural killer T cells, mucosal-associated invariant T (MAIT) cells, ILC3 cells, and B-cells [24].
The role of IL-17 cytokines in asthma is still under investigation. IL-17 cytokines may stimulate epithelial cells and fibroblasts to release neutrophil chemoattractants CXCL1/5/8 and granulocyte–macrophage colony-stimulating factor, which recruit neutrophils to the lungs. Furthermore, IL-17A, but not IL-17F, enhances airway smooth muscle contraction [21], migration [25], and proliferation [26], which facilitates airway hyperresponsiveness (AHR) and airway remodeling, key characteristics of asthma. However, it has been proposed that IL-17 cytokines are important for maintaining the integrity of the epithelium and IL-17 cytokines may play a protective role against asthma [24] (Figure 2).
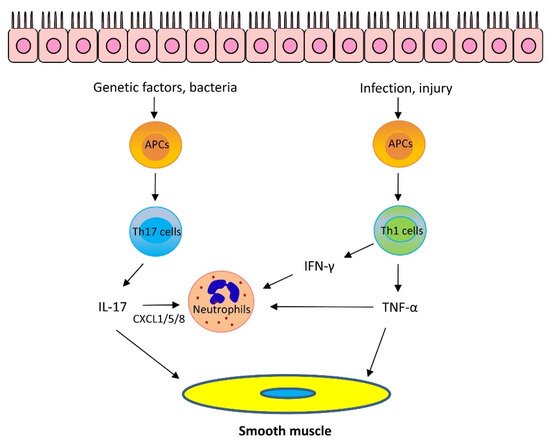
Figure 2. Mechanism of Th2-low asthma. Th17 cytokines: Bacteria promote Th17 cell differentiation via antigen-presenting cells (APCs). Variants in the IL-17 pathway genes also contribute to IL-17 upregulation. IL-17 can stimulate epithelial cells and fibroblasts to release neutrophil chemoattractants CXCL1/5/8 which recruit neutrophils to the lungs. Furthermore, IL-17A enhances airway smooth muscle contraction, migration, and proliferation, which facilitates AHR and airway remodeling, Th1 cytokines: Infection and epithelial injury promote Th1 cell maturation and secrete Th1 cytokines, including TNF-α and IFN-γ. TNF-α synergizes with IL-17 cytokines to promote neutrophil recruitment. Furthermore, TNF-α enhances airway smooth muscle contraction. IFN-γ and TNF-α upregulate Ca2+ signaling in airway smooth muscle and induces AHR. In addition, IFN-γ promotes neutrophil recruitment in the presence of IL-17 cytokines.
2.2. Other Cytokines
It is known that Th1 cells secrete IL-2, interferon-γ (IFN-γ), and lymphotoxin-α and stimulate Th1 immunity, which is characterized by prominent phagocytic activity. However, recent studies suggest that some Th1 cytokines may contribute to asthma pathogenesis. Tumor necrosis factor-α (TNF-α) is a pleiotropic Th1 cytokine, which plays a role in the pathogenesis of inflammatory diseases, including allergy. Sputum TNF-α is elevated in neutrophilic and severe asthma [27]. TNF-α is proposed to synergize with IL-17 cytokines to promote neutrophil recruitment [1][24][1,24]. However, TNF-α may also promote the production of Th2 cytokines, such as IL-4, IL-5, and IL-13 [28]. Furthermore, TNF-α enhances airway smooth muscle contraction, which may contribute to the development of AHR [29] (Figure 2).
IFN-γ, IL-1β, and TNF-α have been shown to upregulate the expression of CD38 (cluster of differentiation 38), also known as cyclic ADP ribose hydrolase in airway smooth muscle cells, which may upregulate intracellular Ca2+ signaling and induce AHR. Knockout (KO) of CD38 reduced AHR in a murine model of asthma [30][31][30,31]. In addition, IFN-γ promotes neutrophil recruitment in the presence of IL-17 cytokines [1][24][1,24].
3. Emerging Mechanisms of Asthma
Asthma has long been viewed as an inflammatory disease. However, there is accumulating evidence to suggest that inflammation-independent processes are also associated with asthma progression. For instance, recent studies demonstrate that protein kinases, adapter proteins, and other molecules contribute to asthma pathogenesis [32][33][34][35][36][37][38][39][40][32,33,34,35,36,37,38,39,40] (Figure 3).
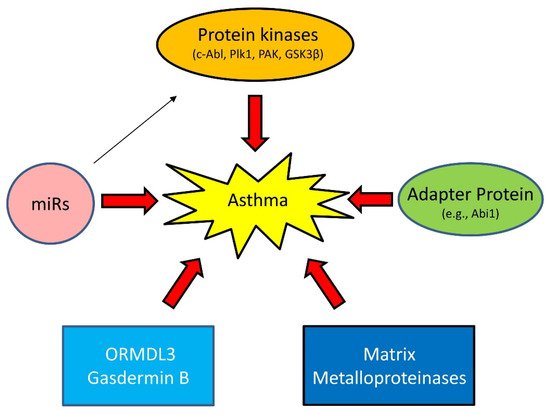
Figure 3. Emerging mechanisms of asthma. Asthma has long been viewed as an inflammatory disease. However, there is accumulating evidence that inflammation-independent processes also contribute to asthma progression. Genetic variance and epigenetics (e.g., miRs) affect expression of proteins, including kinases, adapter protein, ORMDL3, Gasdermin B, and matrix metalloproteinases in lung tissues, which drive asthma progression.
3.1. Proteins Kinases
c-Abl (Abelson tyrosine kinase, Abl, ABL1) is a non-receptor tyrosine kinase that participates in the regulation of smooth muscle contraction, migration, and proliferation [38][41][42][43][44][45][38,41,42,43,44,45]. c-Abl is upregulated in asthmatic human airway smooth muscle (HASM) cells, which is regulated by epigenetic factors [46][47][46,47]. c-Abl KO or inhibition reduces asthma-like phenotypes in animal models of asthma [38][45][38,45]. Furthermore, c-Abl KO or inhibition diminishes Th2 cytokines in experimental asthma [38][45][38,45]. These results suggest that c-Abl is a Th2-regulatory protein rather than a Th2-dependent protein. Intriguingly, treatment with the c-Ab/KIT inhibitor imatinib relieves the symptoms of severe refractory asthma [48].
Polo-like protein kinase 1 (Plk1) is a serine/threonine kinase that plays a role in modulating smooth muscle contraction [37][49][37,49], proliferation [50][51][50,51], migration [50], mitosis [52][53][52,53], and apoptosis [40]. In asthmatic HASM cells, downregulation of miR509 leads to elevated Plk1 [50]. Smooth muscle conditional KO of Plk1 inhibits asthma progression in a murine model of asthma [52]. Plk1 may contribute to airway remodeling via promoting ASM proliferation/migration and inhibiting apoptosis [40][50][52][54][40,50,52,54]. However, Plk1 does not affect Th2 inflammation in experimental asthma [52].
p21-activated kinase (PAK) regulates smooth muscle contraction by modulating the vimentin network and paxillin complexes [54][55][54,55]. Furthermore, a PAK inhibitor or PAK KO protects mice from AHR and airway smooth muscle hyperactivity in vitro [56]. However, it is unclear whether PAK expression and activity are altered in the lungs or serum of asthmatics. Another protein kinase glycogen synthase kinase-3β (GSK-3β) is also linked to asthma pathology. Airway smooth muscle hyperplasia and hypertrophy correlate with GSK-3β phosphorylation in a mouse model of asthma [57]. GSK3 negatively regulates smooth muscle gene expression and hypertrophy. Phosphorylation of GSK3 disinhibits smooth muscle gene expression and promotes ASM hypertrophy and hyperplasia [57][58][57,58].
3.2. Adapter Protein
Abi1 (Abelson interactor 1) is an adapter protein that regulates cell migration [59][60][59,60], smooth muscle contraction [61], and cell proliferation [39]. The human Abi1 gene is localized in the Chromosome 10p21 region. Genome-wide association studies (GWAS) suggest that Chromosome 10p21 is adjacent to a susceptible locus for asthma and related traits [62][63][62,63]. Abi1 is upregulated in asthmatic HASM cells/tissues [39]. Loss-of-function studies suggest that Abi1 contributes to aberrant HASM cell proliferation and asthma phenotype in a murine model of asthma [39].
3.3. MicroRNAs (miRNAs)
miRNAs are evolutionarily conserved, 18–25 nucleotides, noncoding RNA molecules that control gene expression by binding to complementary sequences in the 3′ untranslated regions (3′ UTR) of target mRNAs, which degrade target mRNA and/or repress translation [64]. The levels of miR-203 are downregulated in human asthmatic ASM cells, which disinhibits c-Abl expression and promotes asthma development [46][65][46,65]. Moreover, the expression of miR-509 is lower in human asthmatic ASM cells, which is responsible for the upregulation of Plk1 and asthma progression [47][50][47,50]. miR-25 expression is associated with alterations in ASM cell phenotype, an important process for airway remodeling [66]. miR-144–3p has been shown to be associated with severe corticosteroid-dependent asthma [67].
3.4. Others
ORMDL3 and gasdermin B. GWAS suggest that chromosome 17q21 is linked to asthma [68][69][68,69]. Chromosome 17q21 contains a cluster of genes, including ORMDL3 and gasdermin B (GSDMB) [69]. ORMDL3 may contribute to asthma progression by modulating store-operated calcium entry and lymphocyte activation [70], eosinophil trafficking and activation [71], and sphingolipid homeostasis [72]. Gasdermin B may promote AHR and airway remodeling, without affecting airway inflammation via remodeling-associated gene expression [73].
Matrix Metalloproteinases (MMPs) are calcium-dependent zinc-containing endopeptidases with more than 20 isoforms. MMPs have been linked to asthma, which is isoform dependent [74][75][74,75]. Single-nucleotide polymorphisms (SNPs) in the gene encoding MMP-12 is associated with FEV1 in children and adults with severe asthma [76]. The SNPs in the MMP-12 promoter region increase MMP-12 expression, which may activate macrophages and promote asthma progression [74]. In addition, mast cell tryptase proteolytically activates pro-MMP-1 generated by ASM, which subsequently degrade the extracellular matrix and promote ASM cell growth and airway remodeling [77]. However, MMP-2 appears to have a protective role in asthma. Mice overexpressing human MMP-2 showed a significant reduction in AHR, Th2 cytokines, and IgE compared to their wild-type counterparts [75].