Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Jessie Wu and Version 6 by Jessie Wu.
Arsenic is a chemical element that is toxic, and long-term exposure to it causes cancers such as lung, skin, liver, and bladder cancers. Over 150 million people around the world are affected by arsenic exposure.
- arsenic
- carcinogenesis
- DNA methylation
1. Arsenic and Mechanisms of Arsenic-Induced Carcinogenesis
Arsenic (As), a chemical element, is classified as a toxic metalloid and is associated with various human cancers [1], and its toxicity depends on the molecular form and oxidation state. International Agency for Research on Cancer (IARC) and US Environmental Protection Agency (USEPA) designated arsenic as Group 1 and Group A human carcinogens, respectively [2][3]. Furthermore, it is graded as first on the substance priority list in the Agency for Toxic Substances and Disease Registry (ASTDR), USA (https://www.atsdr.cdc.gov/spl/index.html, accessed on 9 February 2022) [4]. Chronic exposure to dietary arsenic is linked to skin, bladder, liver, and lung cancer [4][5][6][7]. Drinking water contaminated with arsenic has been linked with increased mortality of both noncancerous diseases and cancers in Bangladesh [8]. Both chronic and acute exposure to arsenic is harmful to different tissues and organs in the body, such as alteration in skin pigmentation and hyperkeratosis, peripheral neuropathy, development, cognitive impairments, and cardiovascular diseases.
The contamination of arsenic increases with the finding of newer places [9]. The familiar sources of arsenic exposure include drinking water, food, and inhalation in an industrial work setting. Over 150 million people on the earth are exposed to carcinogenic (10 μg/L) levels of arsenic [9][10], and the majority of these people are affected by drinking water from aquifers contaminated with arsenic. Countries with arsenic concentrations exceeding this carcinogenic level (10 μg/L) in the drinking water include Bangladesh, India, China, Argentina, Mexico, Canada, the USA, and Chile [11]. Arsenic exposure to foods usually occurs by growing crops in the soil contaminated with arsenic and/or irrigating water contaminated with arsenic [12]. Furthermore, NIOSH estimates that approximately 1.5 million workers have been affected by arsenic or arsenic compounds [13].
Arsenic has several states; the most common valence states of arsenic are inorganic AsIII (arsenite) and AsV (arsenate). Inorganic arsenic is very toxic to humans, whereas organic arsenic has low toxicity. AsIII is the highest toxic form because it is more soluble in water than arsenic compounds. It contains a lone electron pair that can engage in chemical bonds [14][15]. Depending on the types of food, arsenic can be found in both inorganic (when combined with oxygen, chlorine, and sulfur, among other elements) and organic forms (when linked with carbon and hydrogen). Inorganic arsenic is typically found in the inorganic form in drinking water, soil, and some terrestrial foods such as rice, as either AsIII or AsV. Inorganic pentavalent arsenic AsV is absorbed by the body through drinking water and uses membrane transporters such as aquaporin and inorganic phosphate transporters (PiT) to enter the cells [16][17]. In the cell, arsenic AsV is converted to the more toxic form arsenite in a glutathione-dependent reaction (GSH), with subsequent methylation to mono-methylated (MMA) and di-methylated arsenicals (DMA), respectively [18][19]. Methylated arsenicals, especially MMAIII, are considered more toxic than inorganic AsIII both in vivo (in hamsters) [20] and in vitro (human cell lines) [21].
The mechanisms by which arsenic induces carcinogenesis are still a point of debate. However, it has been known that arsenical compounds contribute to carcinogenesis by disrupting the signaling cascade, changing gene expression, elevating levels of oxidative stress and inflammation, increasing genotoxic and DNA damage, decreasing DNA repair, inducing cell cycle arrest and apoptosis [19][22][23][24], acting as co-carcinogenesis with other environmental toxicants [25], and alterations of epigenetic regulation. There is also doubt about whether arsenic is genotoxic or not because arsenic does not cause point mutations in standard mutagenicity assays; hence it is considered to be nongenotoxic [26][27]. Although arsenic is viewed as a carcinogen, its non-mutagenic characteristics violet its function in causing genetic alteration. However, a less studied mechanism, but one that is crucial for understanding arsenic-induced carcinogenesis, is the dysregulation of epigenetic modifications. Studies investigating epigenetic regulation changes upon arsenic exposure during the last decade are increasing. The researchers have attempted to explore the role of DNA methylation, histone modification, miRNAs and lncRNAs alteration, mRNA modification, and alternative splicing in arsenic toxicity and carcinogenesis. The present resviearchw will comprehensively discuss the epigenetic regulations involved in gene expression, and their dysregulation is pivotal in arsenic-induced transformation, tumor growth, and angiogenesis. The general scheme of the mechanism of arsenic-induced carcinogenesis is shown as follows (Figure 1).
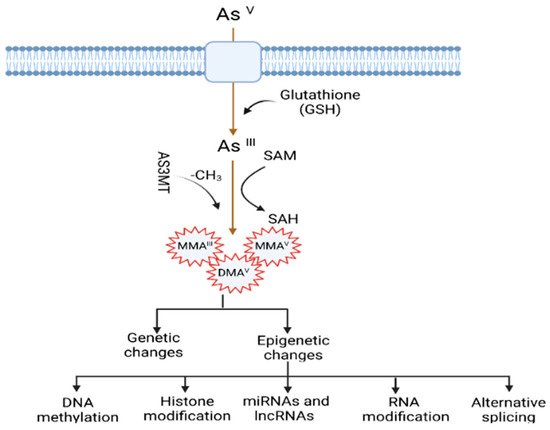
Figure 1. Mechanisms of arsenic-induced carcinogenesis. Arsenic exposure induces carcinogenesis via its biotransformation process, which causes effects on both genetic and epigenetic levels. The biotransformation of arsenic happens via a series of reactions such as reduction, oxidation, and methylation. Pentavalent arsenic (AsV) is reduced to trivalent (AsIII) and then methylated into organic arsenic species with higher carcinogenic potential. Here, S-adenosylmethionine (SAM) acts as a methyl donor, and Glutathione (GSH) and other thiols serve as reducing agents. Epigenetic alterations induced by arsenic exposure include abnormal changes in DNA methylation, histone modification, miRNAs and lncRNAs expression, RNA modification, and alternative splicing.
2. Arsenic-Induced Changes in DNA Methylation
DNA methylation is the inclusion of the methyl group (-CH3) in the 5-carbon on the cytosine residues (5 mC) in CpG (Cytosine-Phosphate-Guanine) and non-CpG (CpA, CpT, and CpC) dinucleotides. The methyl group comes from a methyl donor, generally from S-adenyl methionine (SAM), and this process is mediated by DNA methyl transferases (DNMTs) [28]. CpG dinucleotides are concentrated in CpG islands (short CpG-rich DNA stretches) and regions of repetitive sequences such as centromeric repeats, retrotransposon elements, rDNA, etc. [29][30][31]. In cancers, the changes of methylation status mainly occur within CpG islands, which occupy ~70% of all mammalian promotors. In addition, these islands play an important role in the regulation of transcription, and their general changes have been found during malignant transformation [32][33]. The functional effect of the dysregulation of DNA methylation is context- and spatial-dependent, dynamic, tissue-specific, and trans-generationally heritable [34][35][36]. Generally, gene silencing involves promotor methylation, and constitutive gene expression is associated with gene body methylation [32]. However, the methylation of the gene body may also be found to inactivate repetitive DNA elements within the gene body [35][37] and show dramatic alteration intron-exon boundaries [38]. These complex methylation patterns underline the necessity of DNA methylation profiling to answer biological questions.
Although it is evident that the dysregulation of DNA methylation has been demonstrated in different cancers, people's ur knowledge of the impact of inorganic arsenic (iAs) on DNA methylation is still growing. Methyl transferase (MTs) catalyze the methyl group transfer in the 5-carbon on the cytosine residues (5 mC) in CpG dinucleotides and use SAM, a coenzyme, as a methyl group donor. Long-term exposure to arsenic causes depletion of the SAM by MTs such as AS3MT [arsenic (III) methyl transferase]. Furthermore, arsenic can also control DNMTs and decrease their activities. For example, studies have found that arsenic exposure causes a reduction of mRNA levels and activity of DNMTs [39][40][41].
iAs exposure has been shown to change global DNA methylation in vitro, in animal studies as well as in population studies (Table 1). For instance, a chronic low-dose of iAs exposure induces DNA hypo-methylation in cells [42]. In addition, fish, mice, and rats exposed to iAs exhibit hepatic global DNA hypomethylation [42][43][44]. However, limited studies are available for the human population compared to in vitro and animal studies. A recent study assessed the association between arsenic exposure and global DNA methylation (∼850,000 CpGs) through drinking water among 396 Bangladeshi people who joined the Health Effects of Arsenic Longitudinal Study (HEALS). The study identified 34 CpGs associated with arsenic concentration in the urinary tract and found a positive relationship between higher arsenic concentration and DNA hypomethylation in those CpGs. Among the arsenic-associated CpGs, most of the genes were annotated to the reactive oxygen species (ROS) pathway, tumor necrosis factor-α (TNF-α) signaling, and inflammatory response via nuclear factor kappa B (NF-κB). These are essential hallmarks of cancer and aging [45]. The results are consistent with earlier studies indicating that epigenetic alterations potentially regulate arsenic toxicity [45]. Pilsner et al. showed that iAs exposure led to global hypomethylation of leukocytes in human skin. The authors observed that people with hypo methylation in the peripheral blood lymphocytes (PBL) DNA were prone to skin lesions two years later when they adjusted for age, urinary As, and other factors [46]. A whole-genome microarray-based study showed that the status of DNA methylation changed over time in people who were affected by arsenic-induced skin lesions compared to control in Bangladesh. The study found the top 20 differentially methylated CpG sites. Among these top CpG sites, the methylation percentages increased in 13 CpGs, and decreased in 7 CpGs between baseline and follow-up [47]. Bandyopadhyay et al. evaluated the association of cytogenetic damage by measuring lymphocyte micronucleus (MN) frequency and long interspersed nuclear element-1 (LINE-1) methylation status among children who were exposed to arsenic in the areas of West Bengal. They observed that a high reduction of LINE-1 methylation was associated with MN frequency in exposed children compared to unexposed children, suggesting that LINE-1 methylation is a potential epigenetic marker for arsenic toxicity in individuals [48].
Besides the changes in global DNA methylation status, iAs exposure also causes changes in DNA methylation in specific regions of targeted genes in different cancers [49]. For example, the association between arsenic exposure and hypomethylated or hypermethylated promotors of some genes was found in human skin cancer [50] and bladder cancer [51][52]. The carcinogenesis can occur due to the silence of tumor suppressor genes via hypermethylation [40]. Some studies have found that iAs exposure leads to increased methylation of the promotor for tumor suppressor genes such as p15, p16, p53, and death-associated protein kinase (DAPK) in vitro and in vivo [40][50][51], DNA repair-related genes such as ERCC2, RPA1 in human hepatocytes [53], MLH1 in whole human blood [54], and genes associated with the Wnt pathway like MYC and WNT2B [53]. However, another study involving a human population chronically exposed to arsenic demonstrated hypomethylation at the promoter of the DNA repair gene ERCC2 [55]. Smeester et al. comprehensively studied the status of DNA methylation within CpG islands for more than 14,000 genes among arsenic-exposed individuals with skin lesions and without skin lesions [56]. They identified 183 genes with differentially methylated CpG islands, of which 182 were hyper-methylated in individuals with signs of arsenicosis. Gene enrichment analysis showed that most genes involved cancer-linked pathways via genes such as p53. They also identified an arsenic-methylated tumor suppressorome, a complex of 17 known or putative tumor suppressors silenced in human cancers, which includes hypermethylated genes such as chromosome 11 open reading frame 70 (C11orf70), centromere protein E (CENPE), forkhead box F1 (FOXF1), homeobox B5 (HOXB5), homeobox B9 (HOXB9), hsa-mir-126, SWI/SNF related, matrix associated, actin dependent regulator of chromatin subfamily d member 2 (SMARCD2), T-box brain 1 (TBR1), etc. Chanda et al. showed the hypermethylation of GMDS gene fragments in the peripheral blood leukocyte DNA of individuals exposed to arsenic and with skin cancer. They indicated it as a biomarker for arsenic-induced cancer [57]. The AS3MT gene plays an essential role in the metabolism of arsenic and its toxicological mechanism. Gribble et al. found decreased methylation in the promotor region of AS3MT in an arsenic-exposed area in Arizona [58]. However, no further studies have been performed to investigate the association between skin lesion status and AS3MT promoter methylation to date. On the other hand, carcinogenesis can also occur due to the activation of oncogene genes via hypomethylation. For instance, mice treated with iAs showed hypomethylation of the promoter region of oncogene Hras1 and increased mRNA levels of Hras1 [59], which was consistent with another study showing hypomethylation and increased mRNA levels of Hras1 and c-myc in vitro [60][61]. Arsenic exposure also led to Esr1 gene overexpression via hypomethylation of its promoter region, which is closely related to arsenic-induced hepatocarcinogenesis [44]. However, a recent study by Janasik et al. found hypermethylation of genes promoter of Nuclear factor-erythroid factor 2-related factor 2 (NRF2) and Kelch-like ECH-associated protein 1 (KEAP1) among occupationally arsenic-exposed copper mill workers from Poland [62].
DNA methylation inhibition occurs in a site-specific manner by proteins known as the ten-eleven translocation (TET) enzymes [63][64]. These TET enzymes oxidize 5 mc to 5 hyrdoxymethylcytosine (5 hmc). Disruption of this group of proteins has been shown in different types of cancer. Wang et al. showed that As inhibited the TET-mediated DNA demethylation and subsequently induced the hypermethylation in the promotor region to suppress the antioxidant genes 8-oxoguanine DNA glycosylase (OGG1) and glutathione S-transferase Pi 1 (GSTP1), thus increasing oxidative stress in human bronchial epithelial (HBE) cells in vitro [65]. In another recent study, Domingo-Relloso et al. conducted an epigenome-wide association study (EWAS) to compare the association of different As exposure levels and human blood 5 mc and 5 hmc markers in two diverse populations from the Aragon Workers Health Study (AWHS, Spain) and the Folic Acid and Creatinine Trial (FACT, Bangladesh) [66]. The effect of As on site-specific 5 mC and 5 hmC was measured using the Illumina methylation EPIC array on more than 850,000 CpG sites. They indicated different epigenetic effects for low As exposure in the AWHS population and high As exposure in the FACT population. The differentially methylated (DMP) and hydroxymethylated (DHP) positions were primarily found in distinct genomic sites. For example, they found three DMPs annotated to CLEC12A, a gene that plays a role in inflammation and immune response, which was consistent with previous studies [67]. In addition, they also found one DHP annotated to NPLOC4, a gene that has protein processing function in the endoplasmic reticulum (ER) in the FACT population exposed to a high dose of As. This is invariable to a study that reported a role of As in ER stress-associated protein misfolding and apoptosis [68], for which mechanisms are known to be associated with cardiometabolic diseases and cancer.
In addition, arsenic exposure also causes transgenerational genotoxicity and the alteration of global DNA methylation patterns in the animal model. Parental chronic arsenic exposure led to genotoxic damage (F0–F3), different methylation patterns, changes in physical and reproductive parameters, abnormal morphology in the ovaries (F0 and F1) and testicles (F1–F3), and a decline in the quality of sperm (F0–F3, except F2), suggesting that an individual’s early life disruptions can negatively impact later generations’ health [36]. An association was found between low or high-dose exposure to arsenic during gestation with umbilical cord blood DNA methylation. There was increased DNA methylation in CpG sites of LINE-1 and, to a lesser extent, within the promotor region of p16 [69]. Studies also showed the sex-dependent association between arsenic exposure and cord blood DNA methylation status, and the impact was even more prominent in the boys than in the girls [70].
Arsenic doesn’t induce point mutations but causes deletion mutations and chromosomal instability [40]. One possible mechanism by which arsenical compounds contribute to carcinogenesis is the disruption of normal epigenetic marks at specific loci, which may cause changes in gene expression and carcinogenesis [71][72]. Although arsenic exposure was found to alter methylation levels in global DNA and promoters of some genes, current research is hard to understand due to the complexity and insubstantial information provided in the current studies. Further investigations are necessary to systematically explore DNA methylation on a genome-wide level in cell lines exposed to arsenic and target tissues from well-characterized arsenic-exposed populations or tumor tissues from arsenic-associated cancers. Such studies will assist in elucidating the possible biological effects of arsenic exposure on DNA methylation and carcinogenesis. Arsenic-induced alterations of DNA methylation status and carcinogenesis are summarized in Table 1.
Table 1.
Arsenic-induced alterations of DNA methylation status and carcinogenesis.
| Tissue/Cells | Source of Arsenic | DNA Methylation | References | ||
|---|---|---|---|---|---|
| Global | Gene-Specific | ||||
| Hyper | Hypo | ||||
| Prostate epithelial cell line RWPE-1 | AsIII | Hypo | [73][74] | ||
| HaCaT keratinocytes | AsIII | Hypo | [39] | ||
| TRL 1215 rat liver epithelial cells | AsIII | Hypo | [42] | ||
| Goldfish | AsIII | Hypo | [75] | ||
| Fisher 344 rat | AsIII | Hypo | [43] | ||
| 129/SvJ mice | AsIII | Hypo | [44] | ||
| Blood samples | Drinking water | Hypo | [45] | ||
| Blood samples from skin lesion patients and control | 13 Hyper and 7 hypo methylation of CpG islands | [47] | |||
| Human | Hyper | [76] | |||
| Hypo (in skin lesion patients) | [46] | ||||
| Peripheral blood lymphocyte DNA from skin lesions and non-skin lesions | Drinking water (urine samples) | 182 genes out of 183 hypermethylated; Identified a silenced tumor suppressorome consists of 17 genes |
[56] | ||
| MMAIII | ZHCAN12 and C1QTNF6 | ||||
| Uroepithelial SV-HUC-1 cells | AsIII | DAPK | [77] | ||
| Hamster embryo cells | AsIII | c-myc and Ha-ras | [61] | ||
| TRL 1215 rat liver epithelial cells | AsIII | c-myc | [51] | ||
| C57BL/6J mice | AsIII | c-Ha-ras | [59] | ||
| A/J mice | AsV | p16, RASSF1 | [78] | ||
| C3H mice | AsIII | ERα | [79] | ||
| Blood samples from the people of West Bengal, India | Drinking water | p53 and p21 in skin cancer patients | [50] | ||
| Tissues from arsenic-induced skin lesions (cases) and with no skin lesions (controls) | Drinking water | DAPK and p16 | [80] | ||
| Blood samples from copper mill workers and Non-occupationally exposed healthy controls in Poland | Copper mill (urine) | NRF2 and KEAP1 | [62] | ||
| Blood samples from arsenic-exposed individuals (with and without skin lesions) | Drinking water (water, urine) | MLH1 and MSH2 | [81] | ||
| Samples from bladder tumor | Drinking water (toenail) | RASSF1A and PRSS3 | [52] | ||
| Cord blood lymphocytes | Drinking water (cord blood, nails, and hair) | p53 | [82] | ||
| Blood samples from the West Bengal population and HEK293 cell lines | Drinking water(water, urine), sodium arsenite, AsIII | Increased ERCC2 expression | [55] | ||
| Blood samples from arsenic-exposed individuals (with and without skin lesions) | Drinking water (water, urine) | Increased Tfam and PGC1α expression | [83] | ||
References
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; World Health Organization; International Agency for Research on Cancer. Some Drinking-Water Disinfectants and Contaminants, Including Arsenic; IARC: Lyon, France, 2004; Volume 84, pp. 1–477.
- Bardach, A.E.; Ciapponi, A.; Soto, N.; Chaparro, M.R.; Calderon, M.; Briatore, A.; Cadoppi, N.; Tassara, R.; Litter, M.I. Epidemiology of chronic disease related to arsenic in Argentina: A systematic review. Sci. Total Environ. 2015, 538, 802–816.
- Straif, K.; Benbrahim-Tallaa, L.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens--Part C: Metals, arsenic, dusts, and fibres. Lancet Oncol. 2009, 10, 453–454.
- Chen, C.J.; Kuo, T.L.; Wu, M.M. Arsenic and cancers. Lancet 1988, 1, 414–415.
- Marshall, G.; Ferreccio, C.; Yuan, Y.; Bates, M.N.; Steinmaus, C.; Selvin, S.; Liaw, J.; Smith, A.H. Fifty-year study of lung and bladder cancer mortality in Chile related to arsenic in drinking water. J. Natl. Cancer Inst. 2007, 99, 920–928.
- Smith, A.H.; Hopenhayn-Rich, C.; Bates, M.N.; Goeden, H.M.; Hertz-Picciotto, I.; Duggan, H.M.; Wood, R.; Kosnett, M.J.; Smith, M.T. Cancer risks from arsenic in drinking water. Environ. Health Perspect 1992, 97, 259–267.
- Hopenhayn-Rich, C.; Biggs, M.L.; Fuchs, A.; Bergoglio, R.; Tello, E.E.; Nicolli, H.; Smith, A.H. Bladder cancer mortality associated with arsenic in drinking water in Argentina. Epidemiology 1996, 7, 117–124.
- Sohel, N.; Persson, L.A.; Rahman, M.; Streatfield, P.K.; Yunus, M.; Ekström, E.C.; Vahter, M. Arsenic in drinking water and adult mortality: A population-based cohort study in rural Bangladesh. Epidemiology 2009, 20, 824–830.
- Sanyal, T.; Bhattacharjee, P.; Paul, S.; Bhattacharjee, P. Recent Advances in Arsenic Research: Significance of Differential Susceptibility and Sustainable Strategies for Mitigation. Front. Public Health 2020, 8, 464.
- Ozturk, M.; Metin, M.; Altay, V.; Bhat, R.A.; Ejaz, M.; Gul, A.; Unal, B.T.; Hasanuzzaman, M.; Nibir, L.; Nahar, K.; et al. Arsenic and Human Health: Genotoxicity, Epigenomic Effects, and Cancer Signaling. Biol. Trace Elem. Res. 2022, 200, 988–1001.
- Chakraborti, D.; Rahman, M.M.; Paul, K.; Chowdhury, U.K.; Sengupta, M.K.; Lodh, D.; Chanda, C.R.; Saha, K.C.; Mukherjee, S.C. Arsenic calamity in the Indian subcontinent What lessons have been learned? Talanta 2002, 58, 3–22.
- Chung, J.Y.; Yu, S.D.; Hong, Y.S. Environmental source of arsenic exposure. J. Prev. Med. Public Health 2014, 47, 253–257.
- Stöhrer, G. Arsenic: Opportunity for risk assessment. Arch. Toxicol 1991, 65, 525–531.
- O’Day, P.A. Chemistry and Mineralogy of Arsenic. Elements 2006, 2, 77–83.
- Zampella, G.; Neupane, K.P.; De Gioia, L.; Pecoraro, V.L. The importance of stereochemically active lone pairs for influencing Pb(II) and As(III) protein binding. Chemistry 2012, 18, 2040–2050.
- Wang, Y.; Fang, J.; Leonard, S.S.; Rao, K.M. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1434–1443.
- Hubaux, R.; Becker-Santos, D.D.; Enfield, K.S.; Rowbotham, D.; Lam, S.; Lam, W.L.; Martinez, V.D. Molecular features in arsenic-induced lung tumors. Mol. Cancer 2013, 12, 20.
- Drobna, Z.; Styblo, M.; Thomas, D.J. An Overview of Arsenic Metabolism and Toxicity. Curr Protoc Toxicol 2009, 42, 4–31.
- Davey, J.C.; Nomikos, A.P.; Wungjiranirun, M.; Sherman, J.R.; Ingram, L.; Batki, C.; Lariviere, J.P.; Hamilton, J.W. Arsenic as an endocrine disruptor: Arsenic disrupts retinoic acid receptor-and thyroid hormone receptor-mediated gene regulation and thyroid hormone-mediated amphibian tail metamorphosis. Environ. Health Perspect 2008, 116, 165–172.
- Petrick, J.S.; Jagadish, B.; Mash, E.A.; Aposhian, H.V. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem. Res. Toxicol. 2001, 14, 651–656.
- Styblo, M.; Del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Reed, W.; Wang, C.; Cullen, W.R.; Thomas, D.J. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000, 74, 289–299.
- Chatterjee, A.; Chatterji, U. Arsenic abrogates the estrogen-signaling pathway in the rat uterus. Reprod. Biol. Endocrinol. 2010, 8, 80.
- Cohen, J.M.; Beck, B.D.; Rhomberg, L.R. Historical perspective on the role of cell proliferation in carcinogenesis for DNA-reactive and non-DNA-reactive carcinogens: Arsenic as an example. Toxicology 2021, 456, 152783.
- Hei, T.K.; Filipic, M. Role of oxidative damage in the genotoxicity of arsenic. Free Radic Biol. Med. 2004, 37, 574–581.
- Rossman, T.G.; Uddin, A.N.; Burns, F.J. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl. Pharmacol. 2004, 198, 394–404.
- Rossman, T.G. Mechanism of arsenic carcinogenesis: An integrated approach. Mutat. Res. 2003, 533, 37–65.
- Klein, C.B.; Leszczynska, J.; Hickey, C.; Rossman, T.G. Further evidence against a direct genotoxic mode of action for arsenic-induced cancer. Toxicol. Appl. Pharmacol. 2007, 222, 289–297.
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610.
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21.
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745.
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27.
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734.
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610.
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38.
- Yong, W.S.; Hsu, F.M.; Chen, P.Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin 2016, 9, 26.
- Nava-Rivera, L.E.; Betancourt-Martínez, N.D.; Lozoya-Martínez, R.; Carranza-Rosales, P.; Guzmán-Delgado, N.E.; Carranza-Torres, I.E.; Delgado-Aguirre, H.; Zambrano-Ortíz, J.O.; Morán-Martínez, J. Transgenerational effects in DNA methylation, genotoxicity and reproductive phenotype by chronic arsenic exposure. Sci. Rep. 2021, 11, 8276.
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340.
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331.
- Reichard, J.F.; Schnekenburger, M.; Puga, A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun. 2007, 352, 188–192.
- Ren, X.; McHale, C.M.; Skibola, C.F.; Smith, A.H.; Smith, M.T.; Zhang, L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect 2011, 119, 11–19.
- Chakraborty, A.; Ghosh, S.; Biswas, B.; Pramanik, S.; Nriagu, J.; Bhowmick, S. Epigenetic modifications from arsenic exposure: A comprehensive review. Sci. Total Environ. 2022, 810, 151218.
- Zhao, C.Q.; Young, M.R.; Diwan, B.A.; Coogan, T.P.; Waalkes, M.P. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 10907–10912.
- Uthus, E.O.; Davis, C. Dietary arsenic affects dimethylhydrazine-induced aberrant crypt formation and hepatic global DNA methylation and DNA methyltransferase activity in rats. Biol. Trace Elem. Res. 2005, 103, 133–145.
- Chen, H.; Li, S.; Liu, J.; Diwan, B.A.; Barrett, J.C.; Waalkes, M.P. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: Implications for arsenic hepatocarcinogenesis. Carcinogenesis 2004, 25, 1779–1786.
- Demanelis, K.; Argos, M.; Tong, L.; Shinkle, J.; Sabarinathan, M.; Rakibuz-Zaman, M.; Sarwar, G.; Shahriar, H.; Islam, T.; Rahman, M.; et al. Association of Arsenic Exposure with Whole Blood DNA Methylation: An Epigenome-Wide Study of Bangladeshi Adults. Environ. Health Perspect 2019, 127, 57011.
- Pilsner, J.R.; Liu, X.; Ahsan, H.; Ilievski, V.; Slavkovich, V.; Levy, D.; Factor-Litvak, P.; Graziano, J.H.; Gamble, M.V. Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect 2009, 117, 254–260.
- Seow, W.J.; Kile, M.L.; Baccarelli, A.A.; Pan, W.C.; Byun, H.M.; Mostofa, G.; Quamruzzaman, Q.; Rahman, M.; Lin, X.; Christiani, D.C. Epigenome-wide DNA methylation changes with development of arsenic-induced skin lesions in Bangladesh: A case-control follow-up study. Environ. Mol. Mutagen 2014, 55, 449–456.
- Bandyopadhyay, A.K.; Paul, S.; Adak, S.; Giri, A.K. Reduced LINE-1 methylation is associated with arsenic-induced genotoxic stress in children. Biometals 2016, 29, 731–741.
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428.
- Chanda, S.; Dasgupta, U.B.; Guhamazumder, D.; Gupta, M.; Chaudhuri, U.; Lahiri, S.; Das, S.; Ghosh, N.; Chatterjee, D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol. Sci. 2006, 89, 431–437.
- Chen, W.T.; Hung, W.C.; Kang, W.Y.; Huang, Y.C.; Chai, C.Y. Urothelial carcinomas arising in arsenic-contaminated areas are associated with hypermethylation of the gene promoter of the death-associated protein kinase. Histopathology 2007, 51, 785–792.
- Marsit, C.J.; Karagas, M.R.; Danaee, H.; Liu, M.; Andrew, A.; Schned, A.; Nelson, H.H.; Kelsey, K.T. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 2006, 27, 112–116.
- Miao, Z.; Wu, L.; Lu, M.; Meng, X.; Gao, B.; Qiao, X.; Zhang, W.; Xue, D. Analysis of the transcriptional regulation of cancer-related genes by aberrant DNA methylation of the cis-regulation sites in the promoter region during hepatocyte carcinogenesis caused by arsenic. Oncotarget 2015, 6, 21493–21506.
- Hossain, M.B.; Vahter, M.; Concha, G.; Broberg, K. Environmental arsenic exposure and DNA methylation of the tumor suppressor gene p16 and the DNA repair gene MLH1: Effect of arsenic metabolism and genotype. Metallomics 2012, 4, 1167–1175.
- Paul, S.; Banerjee, N.; Chatterjee, A.; Sau, T.J.; Das, J.K.; Mishra, P.K.; Chakrabarti, P.; Bandyopadhyay, A.; Giri, A.K. Arsenic-induced promoter hypomethylation and over-expression of ERCC2 reduces DNA repair capacity in humans by non-disjunction of the ERCC2-Cdk7 complex. Metallomics 2014, 6, 864–873.
- Smeester, L.; Rager, J.E.; Bailey, K.A.; Guan, X.; Smith, N.; García-Vargas, G.; Del Razo, L.M.; Drobná, Z.; Kelkar, H.; Stýblo, M.; et al. Epigenetic changes in individuals with arsenicosis. Chem. Res. Toxicol. 2011, 24, 165–167.
- Chanda, S.; Dasgupta, U.B.; Mazumder, D.G.; Saha, J.; Gupta, B. Human GMDS gene fragment hypermethylation in chronic high level of arsenic exposure with and without arsenic induced cancer. Springerplus 2013, 2, 557.
- Gribble, M.O.; Tang, W.Y.; Shang, Y.; Pollak, J.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Silbergeld, E.K.; Guallar, E.; Cole, S.A.; et al. Differential methylation of the arsenic (III) methyltransferase promoter according to arsenic exposure. Arch. Toxicol. 2014, 88, 275–282.
- Okoji, R.S.; Yu, R.C.; Maronpot, R.R.; Froines, J.R. Sodium arsenite administration via drinking water increases genome-wide and Ha-ras DNA hypomethylation in methyl-deficient C57BL/6J mice. Carcinogenesis 2002, 23, 777–785.
- Chen, H.; Liu, J.; Zhao, C.Q.; Diwan, B.A.; Merrick, B.A.; Waalkes, M.P. Association of c-myc overexpression and hyperproliferation with arsenite-induced malignant transformation. Toxicol. Appl. Pharmacol. 2001, 175, 260–268.
- Takahashi, M.; Barrett, J.C.; Tsutsui, T. Transformation by inorganic arsenic compounds of normal Syrian hamster embryo cells into a neoplastic state in which they become anchorage-independent and cause tumors in newborn hamsters. Int. J. Cancer 2002, 99, 629–634.
- Janasik, B.; Reszka, E.; Stanislawska, M.; Jablonska, E.; Kuras, R.; Wieczorek, E.; Malachowska, B.; Fendler, W.; Wasowicz, W. Effect of Arsenic Exposure on NRF2-KEAP1 Pathway and Epigenetic Modification. Biol. Trace Elem. Res. 2018, 185, 11–19.
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750.
- Saintilnord, W.N.; Fondufe-Mittendorf, Y. Arsenic-induced epigenetic changes in cancer development. Semin. Cancer Biol. 2021, 76, 195–205.
- Wang, Q.; Wang, W.; Zhang, A. TET-mediated DNA demethylation plays an important role in arsenic-induced HBE cells oxidative stress via regulating promoter methylation of OGG1 and GSTP1. Toxicol. In Vitro 2021, 72, 105075.
- Domingo-Relloso, A.; Bozack, A.; Kiihl, S.; Rodriguez-Hernandez, Z.; Rentero-Garrido, P.; Casasnovas, J.A.; Leon-Latre, M.; Garcia-Barrera, T.; Gomez-Ariza, J.L.; Moreno, B.; et al. Arsenic exposure and human blood DNA methylation and hydroxymethylation profiles in two diverse populations from Bangladesh and Spain. Environ. Res. 2022, 204, 112021.
- Prasad, P.; Sinha, D. Low-level arsenic causes chronic inflammation and suppresses expression of phagocytic receptors. Environ. Sci. Pollut Res. Int. 2017, 24, 11708–11721.
- Zhang, X.Y.; Yang, S.M.; Zhang, H.P.; Yang, Y.; Sun, S.B.; Chang, J.P.; Tao, X.C.; Yang, T.Y.; Liu, C.; Yang, Y.M. Endoplasmic reticulum stress mediates the arsenic trioxide-induced apoptosis in human hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol 2015, 68, 158–165.
- Kile, M.L.; Houseman, E.A.; Baccarelli, A.A.; Quamruzzaman, Q.; Rahman, M.; Mostofa, G.; Cardenas, A.; Wright, R.O.; Christiani, D.C. Effect of prenatal arsenic exposure on DNA methylation and leukocyte subpopulations in cord blood. Epigenetics 2014, 9, 774–782.
- Pilsner, J.R.; Hall, M.N.; Liu, X.; Ilievski, V.; Slavkovich, V.; Levy, D.; Factor-Litvak, P.; Yunus, M.; Rahman, M.; Graziano, J.H.; et al. Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS ONE 2012, 7, e37147.
- Andrew, A.S.; Jewell, D.A.; Mason, R.A.; Whitfield, M.L.; Moore, J.H.; Karagas, M.R. Drinking-water arsenic exposure modulates gene expression in human lymphocytes from a U.S. population. Environ. Health Perspect 2008, 116, 524–531.
- Xie, Y.; Liu, J.; Benbrahim-Tallaa, L.; Ward, J.M.; Logsdon, D.; Diwan, B.A.; Waalkes, M.P. Aberrant DNA methylation and gene expression in livers of newborn mice transplacentally exposed to a hepatocarcinogenic dose of inorganic arsenic. Toxicology 2007, 236, 7–15.
- Coppin, J.F.; Qu, W.; Waalkes, M.P. Interplay between cellular methyl metabolism and adaptive efflux during oncogenic transformation from chronic arsenic exposure in human cells. J. Biol. Chem. 2008, 283, 19342–19350.
- Benbrahim-Tallaa, L.; Waterland, R.A.; Styblo, M.; Achanzar, W.E.; Webber, M.M.; Waalkes, M.P. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: Aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol. Appl. Pharmacol 2005, 206, 288–298.
- Bagnyukova, T.V.; Luzhna, L.I.; Pogribny, I.P.; Lushchak, V.I. Oxidative stress and antioxidant defenses in goldfish liver in response to short-term exposure to arsenite. Environ. Mol. Mutagen 2007, 48, 658–665.
- Pilsner, J.R.; Liu, X.; Ahsan, H.; Ilievski, V.; Slavkovich, V.; Levy, D.; Factor-Litvak, P.; Graziano, J.H.; Gamble, M.V. Genomic methylation of peripheral blood leukocyte DNA: Influences of arsenic and folate in Bangladeshi adults. Am. J. Clin. Nutr. 2007, 86, 1179–1186.
- Chai, C.Y.; Huang, Y.C.; Hung, W.C.; Kang, W.Y.; Chen, W.T. Arsenic salts induced autophagic cell death and hypermethylation of DAPK promoter in SV-40 immortalized human uroepithelial cells. Toxicol Lett. 2007, 173, 48–56.
- Cui, X.; Wakai, T.; Shirai, Y.; Hatakeyama, K.; Hirano, S. Chronic oral exposure to inorganic arsenate interferes with methylation status of p16INK4a and RASSF1A and induces lung cancer in A/J mice. Toxicol Sci. 2006, 91, 372–381.
- Waalkes, M.P.; Liu, J.; Chen, H.; Xie, Y.; Achanzar, W.E.; Zhou, Y.S.; Cheng, M.L.; Diwan, B.A. Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J. Natl. Cancer Inst. 2004, 96, 466–474.
- Banerjee, N.; Paul, S.; Sau, T.J.; Das, J.K.; Bandyopadhyay, A.; Banerjee, S.; Giri, A.K. Epigenetic modifications of DAPK and p16 genes contribute to arsenic-induced skin lesions and nondermatological health effects. Toxicol. Sci. 2013, 135, 300–308.
- Bhattacharjee, P.; Sanyal, T.; Bhattacharjee, S.; Bhattacharjee, P. Epigenetic alteration of mismatch repair genes in the population chronically exposed to arsenic in West Bengal, India. Environ. Res. 2018, 163, 289–296.
- Intarasunanont, P.; Navasumrit, P.; Waraprasit, S.; Chaisatra, K.; Suk, W.A.; Mahidol, C.; Ruchirawat, M. Effects of arsenic exposure on DNA methylation in cord blood samples from newborn babies and in a human lymphoblast cell line. Environ. Health 2012, 11, 31.
- Sanyal, T.; Paul, M.; Bhattacharjee, S.; Bhattacharjee, P. Epigenetic alteration of mitochondrial biogenesis regulatory genes in arsenic exposed individuals (with and without skin lesions) and in skin cancer tissues: A case control study. Chemosphere 2020, 258, 127305.
More