Drug resistance and insensitivity to treatments are the main challenges in breast cancer therapy. Cancer-associated fibroblasts (CAFs) are heterogeneous stromal cells with prevailing roles in cancer development and progression. Epigenetic alterations are essential in regulating CAF activation and heterogeneity. These modifications are druggable targets that can be reversed using pharmacological interventions. CAFs therefore, have remarkable potential as a therapeutic target in breast cancer.
- Breast cancer
- cancer-associated fibroblasts
- heterogeneity
- epigenetic
- post-translational modification
- DNA methylation
- miRNA dysregulation
Please note: Below is an entry draft based on your previous paper, which is written tightly around the entry title. Since it may not be very comprehensive, we kindly invite you to modify it (both title and content can be replaced) according to your extensive expertise. We believe this entry would be beneficial to generate more views for your work. In addition, no worry about the entry format, we will correct it and add references after the entry is online (you can also send a word file to us, and we will help you with submitting).
Definition
Breast cancer is the leading cause of cancer-related mortality in women worldwide. Cancer-associated fibroblasts (CAFs) are a heterogeneous population of cells in the solid tumour microenvironment. These cells are positively linked to breast cancer progression. Breast CAFs can be categorised into distinct subtypes according to their roles in breast carcinogenesis. Epigenetic modifications change gene expression patterns as a consequence of altered chromatin configuration and DNA accessibility to transcriptional machinery, without affecting the primary structure of DNA. Epigenetic dysregulation in breast CAFs may enhance breast cancer cell survival and ultimately lead to therapeutic resistance. A growing body of evidence has described epigenetic modulators that target histones, DNA, and miRNA as a promising approach to treat cancer.
1. Introduction
Targeting stromal components in the tumour microenvironment (TME) is the latest hype in the cancer paradigm. The tight interactions between TME and cancer cells form a protective niche that favour cancer formation, progression and dissemination, thereby leading to chemoresistance and therapeutic failures [1].
The tumour stroma and TME are interchangeable terms that define all non-malignant components in the vicinity of tumours. The tumour stroma consists of cellular components including fibroblasts, adipocytes, lymphoid cells, endothelial cells and mesenchymal stem cells, and non-cellular components such as the extracellular matrix (ECM; Figure 1a). The TME is predominated by cancer-associated fibroblasts (CAFs) which are highly implicated in cancer pathogenesis [2]. CAFs are fibroblast cells within the tumour stroma that exhibit a thin, wavy, and elongated cruciform or stellate-shaped morphology harbouring specific biomarkers [3][4][5][3,4,5]. Compared to normal fibroblasts (NFs), CAFs are larger cells with indented nuclei and possess a more basophilic cytoplasm that contains abundant rough endoplasmic reticulum and free ribosomes, a well-developed Golgi complex, and numerous tension fibres [6]. During a histological examination, CAFs may be identified based on its spindle-shaped morphology and the lack of endothelial, epithelial, and leukocyte markers expression, in addition to the absence of specific cancer mutations [7].
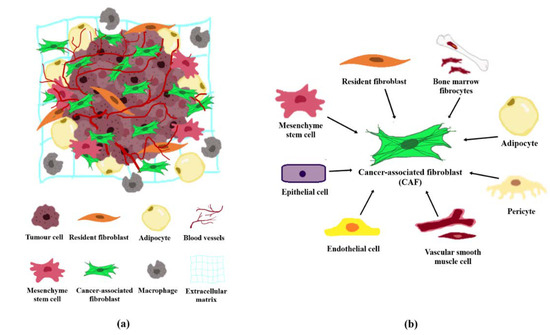
Figure 1.
a
b
CAFs are known to play key roles in cancer progression via secretion of growth factors, cytokines, and chemokines into the TME to support cancer cell proliferation and invasion, ECM remodelling, and increased angiogenesis [4][8][9][4,8,9]. Traditionally, biomarkers used to identify CAFs include alpha-smooth muscle actin (α-SMA), fibroblast-specific protein 1 (FSP1) also known as S100 calcium-binding protein A4 (S100A4), fibroblast activated protein (FAP), platelet-derived growth factor receptor alpha/beta (PDGFRα/β), tenascin-C (TNC), neuron glial antigen (NG2), desmin, cluster of differentiation 90 (CD90) also known asthymocyte differentiation antigen 1 (THY1), podoplanin (PDPN), discoidin domain-containing receptor (DDR2), and vimentin [4][6][10][4,6,10]. However, these biomarkers are expressed in general populations of fibroblast cells and may not be distinct from CAFs. It was only until recently that asporin, collagen XI-α1 (COL11A1), and microfibrillar-associated protein 5 (MFAP5) were described as markers specific to CAFs [4][11][12][13][4,11,12,13].
2. Breast Cancer and CAFs
According to Global Cancer Statistics (GLOBOCAN), breast cancer (BC) is the most common cancer in women worldwide [14]. BC can be categorised into different subtypes based on the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor (HER2) profiles. Broadly, BC sub-categories include the luminal ER-positive [luminal A (ER+, PR+, HER2−) and luminal B (ER+, PR+ with nuclear protein Ki-67 expression, HER2+/−)], HER2 positive (ER−, PR−, HER2+), and the triple-negative (ER−, PR−, HER2−) [15]. Remarkably, breast cancer-associated fibroblasts (BCAFs) are the most prominent cell type that makes up approximately 70% of the breast tumour volume [16]. Although BCAFs lack the biomarkers that are distinguishable from other CAFs, the presence of specific expression patterns in BCAFs (FAP+/α-SMA+/CK−/CD45−) have been frequently observed in the peripheral blood circulating metastatic BC patients [17][18][17,18]. Moreover, Ao and colleagues have reported the presence of circulating BCAFs in 30 of 34 (88%) patients with metastasis and 3 of 13 (23%) patients with localised BC, in a pilot study [17]. Emerging studies have since revealed the multiple features and roles of CAFs in various cancers, including BC [2][6][18][19][20][21][2,6,18,19,20,21].
BCAFs have been reported to alleviate BC tumorigenesis through cancer cell reprogramming metabolic regulation, and ECM remodelling. These alterations can eventually lead to enhanced malignant cell proliferation, tumour invasion, and metastasis [18][20][22][23][18,20,22,23]. According to a study by Mathot and colleagues, the indirect co-culture of various BCAFs (isolated from primary infiltrating ductal or lobular breast carcinoma) with BC cell lines led to the development of aggressive BC phenotypes. These aggressive phenotypes displayed spindle-like morphology with demonstrated reorganised cellular actin [24]. The crosstalk between BC cells and BCAFs encodes a positive feedback loop that facilitates carcinogenesis. BC cells have been reported to activate BCAFs from the surrounding NFs through secretion of platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β). In return, BCAFs may secrete growth factors to fuel tumour growth and confer drug resistance [25].
Compared to mono-cultured BC cells, co-culture of BC cells and BCAFs increased the regulation of FSP1, TGF-β, PDGFβ, fibroblast growth factor 7 (FGF7), interleukins (IL-6 and IL-8), matrix metallopeptidases (MMP2 and MMP11), TIMP metallopeptidase inhibitor 1 (TIMP1), and vascular endothelial growth factor A (VEGFA). These altered regulations subsequently enhanced breast tumour growth, angiogenesis, metastasis, and invasion [26]. The C-X-C motif chemokine ligand 12 (CXCL12) and insulin-like growth factor 1 (IGF1) are CAF-derived cytokines responsible for the pro-survival phosphoinositide 3-kinase-protein kinase B (PI3K-Akt) pathway activation in BC cells. The expression of these cytokines was positively correlated with the predisposition of bone metastasis in triple-negative BCs [27]. Remarkably, BCAFs can also induce endocrine and chemotherapeutic resistance to tamoxifen, doxorubicin, and paclitaxel in BC therapies [18][28][18,28].
3. Reprogramming of NFs into CAFs
NFs are quiescent cells involved in the regulation of ECM turnover to facilitate wound healing, tissue inflammation, fibrosis, as well as tumour growth inhibition [6][18][6,18]. The acquisition of pro-tumorigenic CAFs phenotypes from NFs is associated with altered expression of a large number of genes from the latter [29][30][55,56]. In a comparison study of gene expression between BCAFs and NFs, the MICROMAXTM human cDNA microarray system was selected by Singer and colleagues. They reported BCAFs to exhibit higher expression of genes associated with growth factors and cytokines, intercellular interactions, cell communication, and structure maintenance, as well as proteolytic enzyme regulation [31][57]. In a separate study, Bauer and colleagues used the Affymetrix Human Genome U133 Plus 2.0 and an empirical Bayesian model. In this study, twenty-one genes which were closely related to cell paracrine and intracellular signalling, transcription regulation, cell adhesion and migration, were found upregulated in invasive BCAFs isolated from six patients, when compared to NFs [29][55]. The lack of P53 (TP53), cyclin tumour suppressors including cyclic-dependent kinase inhibitor 1A (CDKN1A, also known as P21), CAV1, as well as phosphatase and tensin homolog (PTEN) protein expression have been reported in BCAFs [20][32][20,58]. The compilation of different regulated genes and proteins in activated BCAFs are summarised in Table 1.
Table 1.
|
Protein/Genes |
Regulation |
Ref. |
|
Growth Factors and Cytokines |
Up |
[31][57] |
|
Granulocyte-macrophage colony-stimulating factor receptor (GMCSFR), Pigment epithelium-differentiation factor (PEDF), Kirsten rat sarcoma viral oncogene homolog protein (K-RAS), Effector cell protease receptor 1 (EPR-1), Hepatoma transmembrane kinase ligand (HTK ligand), Alpha-2-macroglobulin receptor-associated protein (RAP) |
||
|
Cell-Cell Interaction, Communication and Cell Structure Maintenance |
Up |
|
|
Osteopontin (OPN), Transmembrane 4 superfamily protein (SAS), Tapasin (NGS-17), Troponin I fast-twitch isoform (TNNI2) |
||
|
Proteolytic Enzymes Regulation |
Up |
|
|
Dihydrodiol dehydrogenase (DHDH), Protein tyrosine phosphatase (PTP), Hematopoietic consensus tyrosine-lacking kinase (HYL) Pro-cathepsin L (zymogen of proteolytic enzyme cathepsin L), Beta-D-galactosidase (β-gal), Tryptophanyl tRNA synthetase (IFNWRS), Rolipram-sensitive 30, 50-cyclic AMP phosphodiesterase |
||
|
Cell Cycle and Metabolism |
Up |
|
|
Guanine nucleotide-binding protein G(q) subunit alpha (GNAQ), Circadian locomoter output cycles protein kaput (CLOCK), 63-kDa protein kinase related to rat ERK3; also known as mitogen-activated protein kinase 4 (MAPK4), Phospholipase A2 (RASF-A PLA2), Pescadillo homolog protein (PES1), Cell division cycle protein 2 (CDC2), Glutamine-dependent asparagine synthetase (Ts11), Calnexin (CNX) |
||
|
Cell Paracrine and Intracellular Signalling |
Up |
[29][55] |
|
Solute carrier family 24 member 3 (SLC24A3), Insulin like growth factor binding protein 2 (IGFBP2), Tumor necrosis factor ligand superfamily member 4 (TNFSF4), Ras protein specific guanine nucleotide releasing factor 2 (RASGRF2), Polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3), Gap junction protein alpha 5 (GJA5), WNT1 inducible signalling pathway protein 1 (WISP1), V-Ets erythroblastosis virus E26 oncogene homolog (ERG), V-Yes-1 Yamaguchi sarcoma viral related oncogene homolog (LYN) |
||
|
Cell Adhesion and Migration |
Up |
|
|
Transforming growth factor beta 2 (TGF‑β2), ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 5 (ST6GALNAC5), Collagen type X alpha 1 chain (COL10A1), Synaptopodin (SYNPO), Heparin binding EGF like growth factor (HBEGF) |
||
|
ECM Remodelling |
Up |
[33][59] |
|
Plasminogen activator inhibitor 2 (PAI2), Tissue plasminogen activator (PLAT), Matrix metallopeptidase 1 (MMP1), Dickkopf WNT signalling pathway inhibitor 1 (DKK1), Neuregulin 1 (NRG1), Tissue factor pathway inhibitor 2 (TFP12) |
||
|
Steroid Hormone Metabolism |
Down |
|
|
Aldo-keto reductase family 1 member C1 (AKR1C1), Aldo-keto reductase family 1 member C2 (AKR1C2), Phosphatidic acid phosphatase type 2B (PPAP2B), Growth hormone regulated TBC protein 1 (GRTP1) |
||
|
Cell Adhesion and Migration |
Down |
|
|
Slit homolog 3 protein (SLIT3), Osteoblast-specific factor 2 (OSF2), OB-cadherin-1 (CDH11), Cytokine-inducible nuclear protein |
||
|
Tumour Suppressor Genes |
Down |
|
|
Tumour protein P53 (TP53), Cyclin-dependent kinase inhibitor 1A (CDKN1A or P21), Caveolin 1 (CAV1), Phosphatase and tensin homolog (PTEN), Epithelial membrane protein 1 (EMP1) |
Various studies have revealed that disparity in gene expression between CAFs and NFs may be attributed to epigenetic changes [29][34][55,60] or genetic alterations [35][36][61,62]. However, the mechanisms associated with epigenetic regulations underlying BCAFs have not been extensively studied. Hence, this review aims to summarise past and recent findings on the mechanisms involved in epigenetic changes upon BCAFs transformation. This review will also discuss the potential of utilising epigenetic reprogramming in BCAFs as a novel therapeutic approach in targeting BC.
4. Epigenetic Regulation in BCAFs
Epigenetic regulation mechanisms are heritable modifications of chromatin structure or change in gene expression independent of the primary nucleotide sequence [37][63]. Essentially, epigenetic regulation is vital in sustaining the pre-invasive CAFs phenotype and its heterogeneity. Recent studies have shown that the transition of NFs to activated fibroblasts may be regulated epigenetically through a ‘reversible’ or ‘irreversible’ activation process [5]. Fibroblasts that are reversibly activated are known as the normal activated fibroblasts (NAFs). NAFs have high expression of α-SMA and vimentin proteins which corroborate with its function to promote wound healing in healthy tissues. On the other hand, CAFs are the products of irreversible activation in fibroblasts. The epigenetic-driven hyperactivation of fibroblasts often acquires specialised ECM remodelling capability, robust autocrine activation, dynamic immunomodulatory signalling functions, enhanced anti-apoptotic pathways, and proliferative properties. The constitutive activation of this supposed repair pathway eventually leads to cancer fibrosis. However, the comprehensive mechanism governing the activation of CAFs via epigenetics remains elusive. Yet, studies have shown that CAFs could be epigenetically regulated through TME-driven pathways, cancer cells-CAFs crosstalk and CAFs metabolic reprogramming [38][64]. The net effects of the paracrine signalling involving cytokines, exosomes, and metabolites between the cancer cell-CAFs interface also provide a platform for CAFs activation [39][65]. Some of the vital CAF-activating factors released by cancer cells are TGF-β, PDGF, IL-6, IL-1β, epidermal growth factor (EGF), and lysophosphatidic acid (LPA) [10][40][10,66].
The bidirectional crosstalk between tumour cells and CAFs may be facilitated by tumour-derived exosomes (TDEs) [41][42][43][37,40,67]. TDEs are extracellular vesicles that are secreted by the cancer cells or the stroma to transport various bioactive molecules including DNA, microRNAs, proteins, lipids, and metabolites, to facilitate intercellular communication between neighbouring or distant cells [44][45][68,69]. The exosomes released from CAFs can be internalised by the cancer cells to supporttumour progression, whereas the exosomes secreted by the cancer cells could stimulate CAF activation [43][67]. TDEs participate in TME remodelling, angiogenesis, invasion, metastasis, and drug resistance [46][47][70,71]. In BC, TDEs are utilised by BCAFs to transport miRNA, mRNA, and proteins to BC cells for cancer progression [47][48][71,72]. Intriguingly, TDEs are also described as vehicles that transport miRNA-9 (miR-9) from BC cells to breast NFs during BCAF activation in MDA-MD-231 (triple-negative BC) cells [48][72]. The pro-metastatic miRNA-9 could stimulate the transformation of NFs to BCAFs to ultimatelypromote an aggressive BC phenotype [48][72]. Furthermore, the ability of TDEs to transport both TGF-β and IL-6 oncogenic factors may lead to BCAFs transdifferentiation [10][49][10,73].
Alterations within the TME, such as the elevated amount of reactive oxygen species (ROS) or hypoxia, can result in CAFs activation via metabolic reprogramming. The presence of a reactive TME may elevate both hypoxia-inducible factor 1 alpha (HIF-1α) transcription factor and CXCL12 chemokine level; while decreasing CAV1 expression in CAFs [10][50][10,74]. The eventual loss of CAV1 expression in activated CAFs could stimulate TGF-β production and trigger constitutive activation of CAFs [50][74]. Besides, the development of hypoxic CAFs have been linked to enhanced migration and invasion capabilities [51][75]. Hypoxia-induced BCAFs were revealed to possess a high expression of G-protein estrogen receptor (GPR30), which can further support proliferation, metastasis, invasion, and chemoresistance in BC cells [52][53][76,77].
BCAFs can be activated through altered expression of imperative genes and influenced by factors such as the profile of TME and TDEs. The mechanisms underlying epigenetic regulation in BCAFs activation are the post-translational modifications of histone proteins, methylation of DNA, and miRNA regulation. The factors affecting NFs reprogramming and CAFs activation are illustrated in Figure 2.
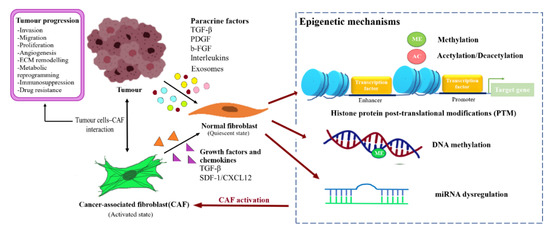
Figure 2. Reprogramming of NFs into CAFs through epigenetic regulation. Fibroblast activating factor secreted by the tumour cells or CAFs caninduced transformation of the normal resident fibroblast into CAFs through epigenetic modulation, and subsequently support tumour progression.
In general, CAFs do not possess genomic alterations but the presence of epigenetic dysregulations including local DNA hypermethylation and global DNA hypomethylation can change the CAFs transcriptional activity, leading to the acquisition of pro-tumorigenic phenotype [54][78]. Secreted factors from either the cancer cells or CAFs can serve as a bridge between BC and CAFs to induce gene or epigenetic modification in both cell types [24]. Yet, the mechanism underlying the epigenetic dysregulation as a result of the crosstalk in cancer remains unknown. The CAF-secreted factors are capable of activating hypermethylation at the cpG island downstream of the transcription start site (TSS) related in BC delineating the significance of epigenetic marks in CAF-dependent BC reprogramming [24].
In-direct co-culture using BCAFs conditioned media on MCF-7 and MDA-MB-231 cell lines resulted in increased expression of TGF-β1, which promoted EMT by up-regulating the expression of IncRNA HOX transcript antisense RNA (HOTAIR) expression in BC. The HOTAIR then mediated H327K tri-methylation of CDK5RAPI and EGR-1 promoters region leading to the elevation of CDK5 protein expression to promote BC invasion and migration [55][79]. Mounting evidence has revealed the importance of TGF-β serves as a major conduit for epigenetic change through BC-BCAFs crosstalk [24][30][55][56][57][58][24,56,79,80,81,82]. The dynamic interactions of histone acetyltransferase (HAT), histone deacetylases (HDACs), histone demethylases (HDMs), histone methyltransferase (HMT) with regulatory-SMAD (R-SMAD) effectors in response to TGF-β signalling are able to modify the gene expression patterns [56][80]. The interaction of P300/CBP and R-SMAD then leads to the acetylation of SMAD protein, to upregulate the genes downstream of the TGF-β signaling axis. On the other hand, the interaction of R-SMAD with HDAC could suppress the TGF-β responsive genes [56][80]. The TGF-β-regulated genes have been associated with various aspects of tumorigenesis including cell proliferation, inflammation, EMT, metastasis, and invasion [59][60][61][83,84,85].
5. Conclusions and Future Perspectives
BCAFs play prevailing roles in cancer progression and are imperative components in the TME, especially in their aptitude to complicate clinical prognosis and treatment response. Considering the implication of BCAFs in BC progression, they, therefore have tremendous potential as a therapeutic target in breast cancer. The ability to discriminate the pro-tumorigenic (F2 subtype) BCAFs from the anti-tumourigenic (F1) subtype is critical, in order to apply the selective inhibition concept by eliminating the cancer-promoting subtype in favour of the cancer-suppression subtype. To date, the distinct BCAF markers associated with F1 and F2 subtypes phenotypes remain unclear. The biological events and mechanisms involved in the programming of the tumour-suppressing subtype-1 are also not well understood. Both BCAF-subtypes may share similar expression of CAF-derived factors or transcriptional signatures, but these molecules may act in opposite manners in different BC, owing to the complexity of the CAF intrinsic properties, TME, and malignant tissue type. In addition to the wealth of knowledge on reported BCAF subpopulations, many candidates remain undiscovered. This opens an avenue for new discovery and investigation into the relationship between various subpopulations and their roles in cancer. The choice of suitable models and techniques that take into consideration the crosstalk within the tumour microenvironment is critical to facilitate the development of CAF-directed therapies [7][62][7,41]. The fundamentals of CAFs recruitment and the dynamics of CAFs-stromal interaction could also be unmasked using the allele and intravital imaging techniques [63][118].
Albeit the variance in epigenetic regulations that give rise to heterogeneity in BCAFs, the underlying mechanisms governing BCAFs have been gradually revealed. However, the mechanisms underlying the epigenetic regulation of unique gene expression patterns and biological behaviour of CAFs remains unclear. The BCAFs heterogeneity and subtypes can exhibit differential expression of genes that affect tumour aggressiveness and immunosuppressive ability, thus investigating their lineages could provide answers to their origins and help in the design of stromal therapeutics. In addition, the distinctive clusters from the heterogeneous BCAF subtypes may also confer contradicting effects (pro- or anti-tumour) toward BC tumour in the TME, causing therapeutic failure. The involvement of BCAF subtypes and the scarcity of unique biomarkers to identify these subpopulations adds a further degree of complexity. Moreover, the lack of distinct yet specific BCAF markers that are exclusively different from other tumour stromal cells also poses a great challenge in targeting BCAFs as a therapeutic modality. The co-existence of similar CAF populations across other cancers sharing similar markers such as S100A4+ and PDPN in breast and CRC cancer [64][65][54,159], suggests that CAFs may possess overlapping functions in different cancers thereby affirming the necessity to redefine CAF classifications using an improved nomenclature system which takes into account specific marker expressions in CAF subpopulations and their functionalities [7]. The single-cell RNA-sequencing (scRNA-seq) and DroNc-seq technique can also be employed to investigate markers that are distinctive to CAF subtypes to address the problem of CAF heterogeneity [66][160]. Nonetheless, targeting several marker combinations instead of a single specific marker on BCAFs could be incorporated in the design of fibroblast-directed therapies to improve treatment efficacy.
Finally, the lack of FDA-approved epigenetic drugs also warrants for concerted efforts between universities and industrial partners to develop new drug candidates for the treatment of solid tumours. The advancement of technologies such as Methylation-Specific PCR (MSP), Combined Bisulfite Restriction Analysis (COBRA), Methylation-sensitive Single Nucleotide Primer Extension (MS-SNuPE) ChIP-seq, and Formaldehyde-assisted Isolation of Regulatory Elements (FAIRE) have also allowed more accurate genome-wide assessment of epigenetic modifications. These high-throughput datasets are beneficial for information-mining towards the identification of more specific biomarkers. On the whole, the issues mentioned above are the critical challenges associated with CAFs biology and epigenetics that deserve further investigation to facilitate the translation of CAF-directed therapy from bench to clinic.