Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Zvonimir Petric.
Monoclonal antibodies (MAbs) have revolutionized the treatment of many chronic inflammatory diseases, including inflammatory bowel disease (IBD). IBD is a term that comprises two quite similar, yet distinctive, disorders—Crohn’s disease (CD) and ulcerative colitis (UC). Two blockbuster MAbs, infliximab (IFX) and adalimumab (ADL), transformed the pharmacological approach of treating CD and UC. However, due to the complex interplay of pharmacology and immunology, MAbs face challenges related to their immunogenicity, effectiveness, and safety.
- monoclonal antibodies
- inflammatory bowel disease
- anti-TNF-α agents
- infliximab (IFX)
- adalimumab (ADL)
1. Introduction
According to the definition, biologic therapy (biologic therapeutic) is a medicine that is made from living organisms (or its products), and is used for the treatment of diseases, as well as for disease prevention or diagnosis [1]. The definitions given by the regulatory agencies in the EU, the European Medicines Agency (EMA), and in the USA, the Food and Drug Administration (FDA), are more or less similar [2,3][2][3]. In 2020, the FDA rephrased its definition by adding that biological product actually refers to all proteins, including any alpha amino acid polymer greater than 40 amino acids [3]. This change, however, has regulatory repercussions without being generally relevant for the clinical practice.
Biopharmaceutical innovation and the implementation of biologic therapy have revolutionized the treatment options for many diseases, from the field of oncology to chronic and inflammatory autoimmune disorders, such as inflammatory bowel disease (IBD) and rheumatoid arthritis (RA), where prior pharmacological attempts with conventional therapy have often been unsuccessful. Hence, it is no surprise that, in the eyes of patients and clinicians, biologic therapeutics are perceived as a game-changing therapeutic modality.
Biologic therapeutics (also called biologics, biologic medicines, biological products, biologics-based medicines, biotherapeutics, biopharmaceuticals, etc.) are well-known as relatively complex molecules produced via a highly sophisticated biotechnological methodology; hence, their high price is unsurprising. Biologics are the fastest-growing therapeutic modality. In 2020, the biologics market was valued to be around EUR 278 billion, while it is expected to reach an astonishing EUR 465 by 2026, according to [4]. Moreover, among the best-selling drugs in 2020, the top-selling was adalimumab (Humira®). Among the top ten drugs, six were monoclonal antibodies (MAbs) [5]. As the global market for biologics is obviously rising, it can be expected that such fast expansion in the coming years will pose big challenges for manufacturers and their production plans if they want to stay at the top in the ever-changing pharmaceutical landscape.
One of the most successful biologics is monoclonal antibodies (MAbs), also referred to as therapeutic antibodies in the case of their multi-indication use. The unique attribute of MAbs is their monospecificity, meaning that they recognize one particular antigenic determinant, i.e., an epitope, on a given molecule. Moreover, as antibodies are secreted by an individual hybridoma, they are completely identical immunoglobulin molecules, which show identical affinity to a target of medical interest, as well as identical physiochemical properties [6].
The nomenclature of MAbs was devised by the World Health Organization (WHO), and follows the International Nonproprietary Nomenclature (INN) (Figure 1), except in the case of muromonab (murine monoclonal antibody) [7].
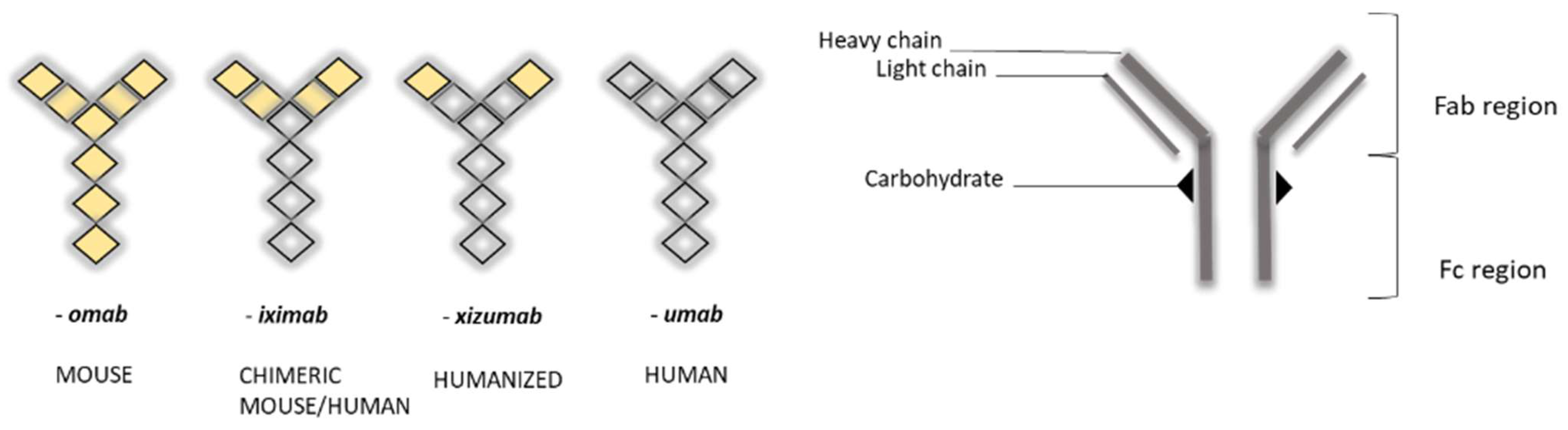
Figure 1. Schematic view of nomenclature for monoclonal antibodies (MAbs) (left) and general representation of their “Y” structure (right) [6]. Note the change in color, representing the differing humanization of antibodies.
In the biopharmaceutical and pharmacological sense, biologics greatly differ from conventional therapy, also known as small-molecule drugs (Table 1). The fundamental differences between these therapeutic modalities, such as their size, chemical structure, physicochemical and biophysical properties, stability, complexity, and specificity, determine the differences in the processes of absorption, distribution, metabolism, and elimination (acronym ADME), i.e., pharmacokinetics (PK), as well as pharmacodynamics (PD). In addition, these differences also influence the way that both therapeutic modalities are manufactured [8].
| Small-Molecule Drugs | Biologics |
|---|---|
| Low molecular weight (<0.5 kDa) | High molecular weight (>2–5 kDa) |
| Small size + lipophilicity allows passage across barriers | Due to its large size, penetration is not expected across barriers |
| Homogenous mixtures | Heterogeneous mixtures, with possible variants |
| Well-defined structure | Structure may not be known (or not well-defined) |
| Physicochemically less complex | Physicochemically very complex |
| Easily synthesized | Made from live cells and organisms |
| Less critical steps in the manufacturing process | Many critical steps in the manufacturing process |
| Very well characterized (methodology is known) | Not easily characterized |
| Stable; heat stable | Not stable; heat sensitive |
| Administered orally | Usually administered parenterally (intravenously, intramuscularly) |
| Relatively short half-life; daily dosing regimen | Longer half-life (days to weeks); monthly dosing regimen |
| High risk for “off-target effects” | High selectivity and specificity for a target |
| Metabolism by liver enzymes—Cytochrome P450 (CYP) | Catabolism (degradation) and limited toxicity |
| Higher risk of drug interactions and toxicity due to CYP | Drug interactions are less common |
| Immunogenicity is not expected | Immunogenicity is a big challenge |
| Treatment is not expensive, i.e., lower costs of development | Treatment is very expensive, i.e., development costs are much higher |
| Longer development cycle | Shorter development cycle |
| Well-defined mechanisms of action | Pleiotropism in pharmacological effects |
| Rigid in terms of structure manipulation | Structure manipulation is possible and can offer an enhancement of pharmacological properties |
2. General Concepts of Pharmacokinetics (PK) and Pharmacodynamics (PD) Related to MAbs
MAbs are 150 kDa immunoglobulin G (IgG) monoclonal antibodies, composed of two heavy chains and two light chains, which are linked by disulfide bonds, and which join to form a molecule resembling the letter “Y” (Figure 1). Tips of the “Y” (i.e., heavy + light chains) are called the variable region, while the stem portion of the “Y” (heavy + heavy chains) is called the constant region. Variable regions comprise an antigen-binding fragment (Fab), while constant regions comprise a fragment crystallizable (Fc) region. The Fab region binds to receptors on the cell’s surface, such as Fcγ receptors (FcγR) and neonatal Fc receptors (FcRn) [10,11][10][11].
The general pharmacological function of MAbs, for example, antagonism against tumor necrosis factor-alpha (TNF-α) cytokines, is dependent on the selective binding of the antibody to the target of interest (antigen) through variable regions. In addition to determining the antibody specificity of an antigen, variable regions also determine the potency of MAbs. On the other hand, the constant region impacts the functional effects of MAbs, such as its developability (i.e., biophysical properties), immunogenicity (i.e., ability to provoke an immune reaction), and effector functions (i.e., binding to receptors and PD). MAbs can also have post-translational modifications, such as amino acid and carbohydrate (glycosylation) modifications. Even a slight change in the constant (or variable) region can have a big and unpredictable impact on the clinical pharmacology of MAbs, meaning that both PK (ADME) and PD (efficacy, effectiveness, and safety) can be altered [10,11][10][11].
As proteins, MAbs have a very low oral bioavailability, poor gastrointestinal stability, and poor lipophilicity, which makes them unsuitable for oral administration. Their apparent volume of distribution is considered to be relatively small and often limited to the circulatory tissue. In a steady state, typical values of the apparent volume of distribution (Vd) are within the range of 3.5–7 L, which indicates the limited distribution of MAbs to vascular and interstitial spaces [9].
The transfer of MAbs from plasma to interstitial space depends on the convective transport (as opposed to diffusion seen with small-molecule drugs), while the rate is determined by capillary permeability. Convection depends on the hydrostatic and osmotic pressure gradients between blood and tissue, but also on the vascular endothelium containing pores, which differ in amount and size. Some tissues may have a more “leaky” endothelium, while capillaries in the brain and their endothelial cells are actually impermeable, meaning that concentrations of MAbs in the brain are less than 1% relative to plasma concentrations [12].
It is also important to mention that MAbs administered via extravascular routes, i.e., intramuscularly (i.m.) or subcutaneously (s.c.), will have a rate of absorption dependent on the convective transport and lymph flow [13,14][13][14]. While the lymph volume can influence the apparent volume of distribution in a steady state [14], it can also be stated that the distribution of MAbs is relatively fast, while elimination (by either excretion or catabolism) is relatively slow [9,13][9][13].
Due to their large size, MAbs are not eliminated by the kidneys in normal situations, while biliary excretion is also not considered to be relevant as the number of MAbs eliminated in this way is very small. Hence, the main elimination of MAbs is facilitated by proteolytic catabolism. Catabolism is mediated via lysosomal degradation (to amino acids) after the uptake of the antibody into cells by two mechanisms. The first uptake mechanism is pinocytosis, a form of unspecific fluid-phase endocytosis, which takes place on the vascular monolayer of endothelial cells. Pinocytosis is not limited to any particular organ or tissue, but instead occurs throughout the body where rich capillary beds are located, i.e., endothelial cells (liver, muscle tissue, skin, gastrointestinal tract, etc.) [12,13,15][12][13][15]. The second uptake mechanism leading to MAb elimination is receptor-mediated endocytosis, where an MAb’s Fc domain interacts with Fc cell receptors (FcγR), leading to endocytotic internalization, and the subsequent inactivation of MAbs (via lysosomal degradation). Various types of immune cells, such as monocytes, macrophages, natural killer (NK) cells, and dendritic cells, express FcγR on their surface membrane [12,16][12][16]. However, one additional interaction is related to receptor-mediated endocytosis, which implies the Fab-binding domain of the antibody to its specific target, i.e., epitope. This is known as a specific clearance pathway of MAbs, and is often referred to as target-mediated drug disposition (TMDD) [9,13,17][9][13][17].
TMDD is considered to be a PK, i.e., drug distribution, phenomenon. It has a lower elimination capacity compared to unspecific pinocytosis and, thus, can be saturable (contrary to an unspecific pinocytosis clearance mechanism that typically shows a linear behavior within the approved therapeutic dosage range). An in-depth explanation of TMDD is beyond the scope here; only its general relevance is presented. In short, TMDD occurs due to a very high affinity and very high binding specificity of the drug for its relatively low-capacity (i.e., low-density) pharmacological target. This phenomenon can be viewed as an example of how PD impacts PK (usually PK impacts PD), and as such, it is relevant to the disposition of biologics, contrary to common belief, as well as for some small-molecule drugs. TMDD could lead to the increased elimination of a drug due to the fact that the drug–target complex molecules can become endocytosed and degraded [13,18,19][13][18][19]. Hence, drugs cleared primarily via TMDD will show dose-dependent nonlinear elimination (even at therapeutic concentrations for some drugs), so TMDD can be considered as an important contributing factor for drug elimination. However, due to the generally high therapeutic concentrations of MAbs used in the clinical setting, TMDD will not usually be the main factor that contributes to increased drug clearance, as there will be the sufficient fraction of a drug (unbound), compared to the fraction (bound, i.e., captured) on the target (receptor). As an example, antibodies against soluble antigens, e.g., tumor necrosis factor-alpha: TNF-α, infliximab (IFX), and adalimumab (ADL), are administered in a high dose and display linear elimination within that therapeutic range [9,13][9][13]. In conclusion, the rate of drug elimination mediated through TMDD will mainly depend on the drug dose, target capacity (density), drug affinity, binding specificity, and the rate of catabolism [13].
Other molecular aspects of pharmacology, which add an additional layer of complexity to the PKPD properties of MAbs, are target turnover rate, changes in the patterns of glycosylation, off-target binding, immunogenicity (i.e., generation of anti-drug antibodies: ADAs) and the FcRn-mediated recycling of MAbs. Due to immunogenicity, i.e., antibody–ADA immune complexes, one can also expect changes in antibody disposition, such as increased clearance and reduced half-life [9]. On the other hand, FcRn-mediated recycling serves as a salvage pathway for MAbs, as it protects antibodies from lysosomal degradation and, thus, it partially counteracts the clearance process. Despite being a capacity-limited process (such as TMDD), FcRn-mediated recycling has a very important PKPD consequence, which is the prolongation of elimination half-life and, consequently, a longer duration of pharmacological effects [20]. Hence, FcRn-mediated recycling can be exploited as a prospective tool for improving the pharmacological properties of antibodies. On the other hand, blocking the FcRn activity was shown to be a good strategy for the treatment of myasthenia gravis. Currently, nipocalimab (anti-FcRn monoclonal antibody) is under clinical trials in adults (phase III) and children (phase II) [21,22][21][22]. Similarly, efgartigimod alfa (antibody Fc fragment) is currently expected to be approved in the EU for the treatment of generalized myasthenia gravis [23]. It is worth mentioning how the expression of Fc receptors in different pathologies can result in a variety of immunological responses, e.g., autoimmunity, inflammation, or allergies. Additionally, the therapeutic effectiveness of MAbs is found to be related to the genetic variants of Fc receptors in individuals [24]. This also means that in IBD, the dysregulation of FcR signaling [25] could have a positive (or negative) influence on the clinical response. In order to be “druggable” enough, the MAb drug target should be easily available and tissue-specific, while, at the same time, maintaining a low receptor turnover rate and low density. The latter properties offer less frequent dosing, or using a drug in lower amounts [26]. In addition to the previously described molecular PKPD complexities related to MAbs, there are also patient-related complexities, which cause interindividual variability in PK, in turn affecting PD. These differences are mostly related to age, pharmacogenetic profile (genetic polymorphisms), concomitant medications, immunogenicity (ADAs), and disease/health status [9,14][9][14]. Hence, as the knowledge on PKPD, inter-patient variability, and underlying pathology is still limited, it is important to bear in mind their joint influence on the pharmacological success of MAbs [14,17][14][17]. Therefore, clinicians often use biomarkers and clinical endpoints as a surrogate for pharmacological success. For example, in the case of IBD, the serum level of C-reactive protein (CRP), or a fecal calprotectin, and the status of mucosal healing are of great help in monitoring disease progression and evaluating the success of pharmacological intervention [9,27,28,29][9][27][28][29].
3. Inflammatory Bowel Disease (IBD)
IBD is an umbrella term, which is mainly used to describe a group of contrasting yet related intestinal disorders: Crohn’s disease (CD) and ulcerative colitis (UC). Both disorders are characterized by non-infectious chronic relapsing episodes of inflammation of the gastrointestinal tract, probably caused by a dysregulation of immune response to the gut microbiome in genetically susceptible individuals [30,31][30][31]. As the etiology and pathophysiology of IBD is puzzling (Figure 2), so far, it has been established that genetic risk factors, environmental factors, lifestyle, mucosal immunity via the intestinal barrier, and the gut microbiome (intestinal dysbiosis) all play a role in the development of the disease. Despite the knowledge on the interplay of these factors, the health burden of IBD is still globally rising. In 2017, according to sources, the number of cases worldwide was 6.8 million, and currently, around 7 million people are living with IBD worldwide [32,33][32][33]. Additionally, the impact of such a burden on the health system in the next few years, especially if we consider the global trends in aging of the population, may become cumbersome.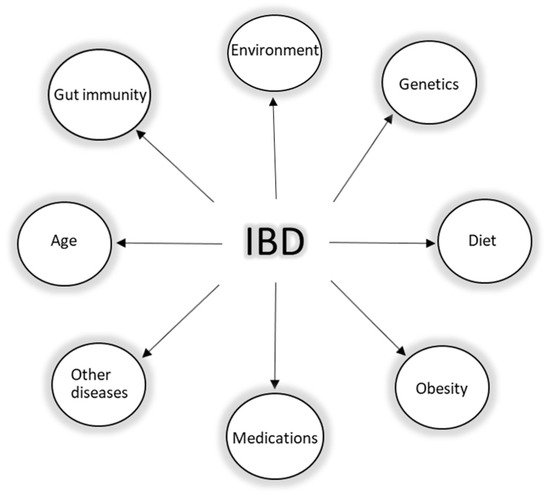
4. Short Immunological Background of IBD
In the healthy gut, Toll-like receptors (TLRs), as pathogen-sensitive innate immune receptors found on monocytes, macrophages, dendritic cells, and epithelial cells, help to maintain the intestinal epithelial barrier. This protective mechanism involves nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which triggers the expression of inflammatory molecules such as TNF-𝛼 and other chemokines. However, in patients with IBD, as barrier function is impaired, TLR signaling is hyperactivated and, consequently, the expression of TNF-α and IL-1, IL-2, IL-6, and IL-12 is elevated [34,50][34][40]. Currently, it is well-established that for the development of IBD, both innate and adaptive (or acquired) immune responses need to be engaged (Figure 3). The innate immune response includes the same cells in CD and UC. Hence, IBD studies show similar increases in macrophages and dendritic cells with the increase in pro-inflammatory cytokines such as TNF-𝛼, a key player in IBD, and others, such as interleukin 1 (IL1), IL-12, and IL-6. On the contrary, the adaptive immune response has a completely different pathway in CD, compared to UC. The inflammation in CD is mediated via the T helper type 1 and T helper type 17 cell-mediated cytokine profile (Th1 and Th17). The inflammation in UC is mediated via natural killer T cells (NK cells) and T helper type 2 cell-mediated cytokine profile (Th2) (Figure 3) [34,50][34][40].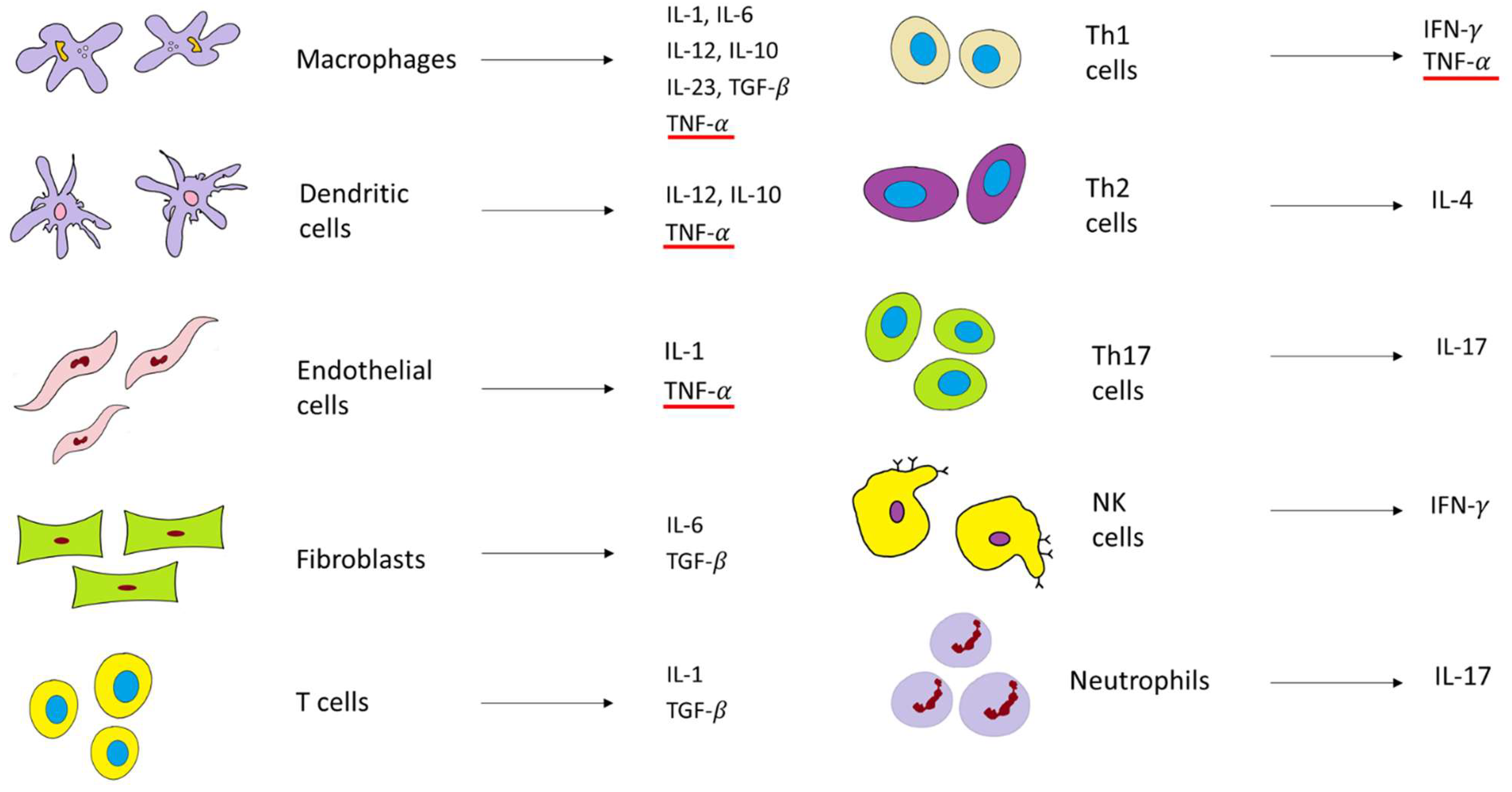
5. Pharmacological Armamentarium of IBD: Targeting TNF-α with Anti-TNF-α Agents—IFX and ADL
Some of the main proinflammatory cytokines include TNF-α, IL-1, and IL-6 (Figure 3). TNF-α is considered to be at the top of the inflammatory cascade and acts as a key player in IBD pathogenesis [50][40]. In healthy (physiological) conditions, as previously stated, TNF-α is a beneficial immune mediator that is responsible for maintaining balanced gut immune homeostasis. However, in the inflammatory state, TNF-α is produced relatively quickly (within one hour) compared to other proinflammatory cytokines. Moreover, TNF-𝛼 has a high potency, as it binds to the receptors with a very high affinity [51,52][41][42]. As it is a transmembrane protein (tm) and expressed on the cell surface, tmTNF-α (also known as mTNF-α) is cleaved by a metalloproteinase, which liberates another form of TNF known as soluble TNF-α (sTNF-α). sTNF-α can be found (and measured) as a homotrimer circulating in the blood. Both mTNF-α and sTNF-α are bound to transmembrane receptor molecules p55/p60 (also known as TNFR1) and p75/p80 (also known as TNFR2), which can also exist in their soluble forms. mTNF-α is a ligand for both these receptors, and their overexpression is additionally upregulated by interferons [53][43]. The binding of TNF-α to receptors forms TNF–TNFR complexes and leads to the overexpression of inflammatory cytokines, cell apoptosis, and necrosis, or alternatively, cell survival, depending on the signaling cascade. One interesting phenomenon related to TNF-α is the possibility of autoupregulation and the creation of a positive pro-inflammatory feedback loop, which further amplifies the inflammatory process [54][44]. Therefore, the concept of the pharmacological targeting of this pleiotropic cytokine [55][45] was a revolutionary step in the early 1990s, when the first experiments confirmed the proof of concept [56,57][46][47]. A few years later, the pharmacological armamentarium of IBD, in addition to conventional therapy, was supplemented by IFX, approved for medical use by the FDA in 1998, while the approval of ADL followed four years later. IFX is a chimeric (human–murine) monoclonal IgG1 anti-TNF-α antibody, while ADL is a fully human monoclonal IgG1 anti-TNF-α antibody (Figure 4) [58,59][48][49].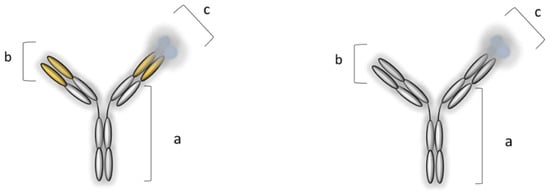
Figure 4. Monoclonal IgG1 anti-TNF-α antibodies (MAbs): infliximab, IFX (on the left), and adalimumab, ADL (on the right). IFX: (a) Human IgG1 constant region, (b) mouse antigen-binding variable region, and (c) homotrimer of TNF-α; ADL: (a) human IgG1 constant region, (b) human antigen-binding variable region, and (c) homotrimer of TNF-α [6,60][6][50].
| IFX | ADL |
|---|---|
| Crohn’s disease Ulcerative colitis Pediatric Crohn’s disease Pediatric ulcerative colitis Rheumatoid arthritis Ankylosing spondylitis Psoriatic arthritis Psoriasis |
Crohn’s disease |
88,93][68][78][83] (in healthy subjects). * denotes the minimum post-induction C trough concentrations of patients with IBD suggested to be associated with an increased likelihood of mucosal healing at week 14 for IFX, and at week 4 for ADL [94][84].
| Anti-TNF-α Agent |
Dose | Route | Cmax µg/mL |
Ctrough * µg/mL |
Tmax Days |
Clearance mL/h |
Half-Life Days |
Vd L |
F % |
AUC µg *h/mL |
|---|---|---|---|---|---|---|---|---|---|---|
| Ulcerative colitis | Pediatric Crohn’s disease Rheumatoid arthritis Juvenile idiopathic arthritis Polyarticular juvenile idiopathic arthritis Active enthesitis-related arthritis Psoriatic arthritis Plaque psoriasis Pediatric plaque psoriasis Axial spondyloarthritis Hidradenitis suppurativa Uveitis Pediatric uveitis Panuveitis |
|||||||||
| IFX | 5 mg/kg | i.v. | 126.2 | >7 | 0.0875 | 11 | 14.1 | 4.8 | 100% | 37 022 |
| Behcet’s disease Pyoderma gangrenosum Hidradenitis suppurativa Graft versus host disease Sjogren’s syndrome Uveitis Kawasaki disease |
Behcet’s disease Pyoderma gangrenosum Alopecia areata Pemphigus Sarcoidosis Wegener’s granulomatosis |
Table 4. Typical pharmacokinetic parameters after single dose of infliximab (IFX) [74,93][64][83] and adalimumab (ADL) [78,
| ADL | 40 mg | s.c. | 3.6 | >7 | 7.9 | 16 | 14.5 | 7.9 | 64% | 2167 |
i.v.—intravenous route; s.c.—subcutaneous route; Cmax—maximum plasma concentration; Tmax—time to reach maximum concentration; Vd—apparent volume of distribution; F—bioavailability; AUC—area under the curve.
Table 5. Differences in routes of administration and dosing of infliximab (IFX) and adalimumab (ADL) in CD and UC [78,92].
Differences in routes of administration and dosing of infliximab (IFX) and adalimumab (ADL) in CD and UC [68][82].
| Biologics | Route | Induction Dose (CD and UC) | Maintenance Dose (CD and UC) |
|---|---|---|---|
| IFX | i.v. |
|
|
| ADL | s.c. |
or
|
* Higher dose is recommended inthe case of unsustained response to IFX ** Initial start on day 29 |
i.v.—intravenous route; s.c.—subcutaneous route; CD—Crohn’s disease; UC—ulcerative colitis.
References
- National Cancer Institute: Dictionary. Biological Drug. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/biological-drug (accessed on 27 June 2022).
- European Medicines Agency. EMA Glossary. Available online: https://www.ema.europa.eu/en/glossary/biological-medicine (accessed on 27 June 2022).
- U.S. Food and Drug Administration (FDA). Definition of the Term “Biological Product”. Available online: https://www.federalregister.gov/documents/2020/02/21/2020-03505/definition-of-the-term-biological-product (accessed on 27 June 2022).
- Mordor Intelligence. Biologics Market–Growth, Trends, COVID-19, and Forecasts (2022–2027). Available online: https://www.mordorintelligence.com/industry-reports/biologics-market (accessed on 27 June 2022).
- Drug Discovery and Development Trends. 50 of 2020’s Best-Selling Pharmaceuticals. Available online: https://www.drugdiscoverytrends.com/50-of-2020s-best-selling-pharmaceuticals (accessed on 27 June 2022).
- Moldenhauer, G. Selection Strategies I: Monoclonal Antibodies. In Handbook of Therapeutic Antibodies; Wiley Online Library: Hoboken, NJ, USA, 2007; pp. 18–44.
- World Health Organisation (WHO). Revised Monoclonal Antibody (mAb) Nomenclature Scheme. Available online: https://www.who.int/medicines/services/inn/Revised_mAb_nomenclature_scheme.pdf (accessed on 27 June 2022).
- Mahler, S. Safety of biologics therapy: Monoclonal antibodies, cytokines, fusion proteins, hormones, enzymes, coagulation proteins, vaccines, botulinum toxins. MAbs 2017, 9, 885–888.
- Deng, R.; Jin, F.; Prabhu, S.; Iyer, S. Monoclonal antibodies: What are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert. Opin. Drug Metab. Toxicol. 2012, 8, 141–160.
- Ovacik, M.; Lin, K. Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin. Transl. Sci. 2018, 11, 540–552.
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103.
- Temrikar, Z.H.; Suryawanshi, S.; Meibohm, B. Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr. Drugs 2020, 22, 199–216.
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588.
- Gill, K.L.; Machavaram, K.K.; Rose, R.H.; Chetty, M. Potential Sources of Inter-Subject Variability in Monoclonal Antibody Pharmacokinetics. Clin. Pharmacokinet. 2016, 55, 789–805.
- Wright, A.; Sato, Y.; Okada, T.; Chang, K.H.; Endo, T.; Morrison, S.L. In vivo trafficking and catabolism of IgG1 antibodies with Fc associated carbohydrates of differing structure. Glycobiology 2000, 10, 1347–1355.
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393.
- Kamath, A.V. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov. Today Technol. 2016, 21–22, 75–83.
- An, G. Concept of Pharmacologic Target-Mediated Drug Disposition in Large-Molecule and Small-Molecule Compounds. J. Clin. Pharmacol. 2020, 60, 149–163.
- Di Paolo, A.; Luci, G. Personalized Medicine of Monoclonal Antibodies in Inflammatory Bowel Disease: Pharmacogenetics, Therapeutic Drug Monitoring, and Beyond. Front. Pharmacol. 2020, 11, 610806.
- Qi, T.; Cao, Y. In Translation: FcRn across the Therapeutic Spectrum. Int. J. Mol. Sci. 2021, 22, 3048.
- Clinical Trials. A Study of Nipocalimab Administered to Adults with Generalized Myasthenia Gravis. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04951622 (accessed on 27 June 2022).
- Clinical Trials. A Study of Nipocalimab in Children Aged 2 to Less than 18 Years with Generalized Myasthenia Gravis. Available online: https://clinicaltrials.gov/ct2/show/NCT05265273 (accessed on 27 June 2022).
- European Medicines Agency. Vyvgart–Efgartigimod Alfa. Available online: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/vyvgart (accessed on 27 June 2022).
- Kim, J.; Lee, J.Y.; Kim, H.G.; Kwak, M.W.; Kang, T.H. Fc Receptor Variants and Disease: A Crucial Factor to Consider in the Antibody Therapeutics in Clinic. Int. J. Mol. Sci. 2021, 22, 9489.
- Castro-Dopico, T.; Clatworthy, M.R. IgG and Fcγ Receptors in Intestinal Immunity and Inflammation. Front. Immunol. 2019, 10, 805.
- Strohl, W.R.; Strohl, L.M. (Eds.) Monoclonal antibody targets and mechanisms of action. In Therapeutic Antibody Engineering; Woodhead Publishing: Sawston, UK, 2012; pp. 163–595.
- Cholapranee, A.; Hazlewood, G.S.; Kaplan, G.G.; Peyrin-Biroulet, L.; Ananthakrishnan, A.N. Systematic review with meta-analysis: Comparative efficacy of biologics for induction and maintenance of mucosal healing in Crohn’s disease and ulcerative colitis controlled trials. Aliment. Pharmacol. Ther. 2017, 45, 1291–1302.
- Dragoni, G.; Innocenti, T.; Galli, A. Biomarkers of Inflammation in Inflammatory Bowel Disease: How Long before Abandoning Single-Marker Approaches? Dig. Dis. 2021, 39, 190–203.
- Sechidis, K.; Papangelou, K.; Metcalfe, P.D.; Svensson, D.; Weatherall, J.; Brown, G. Distinguishing prognostic and predictive biomarkers: An information theoretic approach. Bioinformatics 2018, 34, 3365–3376.
- Faye, A.S.; Colombel, J.F. Aging and IBD: A New Challenge for Clinicians and Researchers. Inflamm. Bowel. Dis. 2022, 28, 126–132.
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317.
- Molodecky, N.A.; Kaplan, G.G. Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. 2010, 6, 339–346.
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30.
- Brajdić, A.; Mijandrušić-Sinčić, B. Insights to the Ethiopathogenesis of the Inflammatory Bowel Disease. In Inflammatory Bowel Disease; Szabo, I., Ed.; IntechOpen Book Series; IntechOpen: London, UK, 2012; p. 292.
- Coward, S.; Clement, F.; Benchimol, E.I.; Bernstein, C.N.; Avina-Zubieta, J.A.; Bitton, A.; Carroll, M.W.; Hazlewood, G.; Jacobson, K.; Jelinski, S.; et al. Past and Future Burden of Inflammatory Bowel Diseases Based on Modeling of Population-Based Data. Gastroenterology 2019, 156, 1345–1353.e1344.
- Stevens, H.; Huys, I. Innovative Approaches to Increase Access to Medicines in Developing Countries. Front. Med. 2017, 4, 218.
- Rajbhandari, R.; Blakemore, S.; Gupta, N.; Adler, A.J.; Noble, C.A.; Mannan, S.; Nikolli, K.; Yih, A.; Joshi, S.; Bukhman, G. Crohn’s disease in low and lower-middle income countries: A scoping review. World J. Gastroenterol. 2020, 26, 6891–6908.
- Falloon, K.; Lazarev, M. A Primer on IBD: Phenotypes, Diagnosis, Treatment, and Clinical Challenges. In Molecular Genetics of Inflammatory Bowel Disease; Hedin, C., Rioux, J.D., D’Amato, M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 3–24.
- Passarella, A.; Grewal, P.; Vrabie, R. Diagnosis and Monitoring in Inflammatory Bowel Disease: Who, When, Where, and How. In Inflammatory Bowel Disease: Pathogenesis, Diagnosis and Management; Rajapakse, R., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 25–59.
- Silva, F.A.; Rodrigues, B.L.; Ayrizono, M.L.; Leal, R.F. The Immunological Basis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 2097274.
- Moreland, L.W. Inhibitors of tumor necrosis factor: New treatment options for rheumatoid arthritis. Clevel. Clin. J. Med. 1999, 66, 367–374.
- Moreland, L.W.; Emery, P. TNF-Inhibition in the Treatment of Rheumatoid Arthritis; CRC Press: Boca Raton, FL, USA, 2004.
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472.
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784.
- Alam, M.S.; Otsuka, S.; Wong, N.; Abbasi, A.; Gaida, M.M.; Fan, Y.; Meerzaman, D.; Ashwell, J.D. TNF plays a crucial role in inflammation by signaling via T cell TNFR2. Proc. Natl. Acad. Sci. USA 2021, 118, e2109972118.
- Widmer, M.B.; Fanslow, W.C.; Jacobs, C.A.; Mohler, K.M.; Lynch, D.H.; Grabstein, K.H.; Maliszewski, C.R. Soluble cytokine receptors as immunosuppressants. Int. J. Cell Cloning 1991, 9, 222.
- Jacobs, C.A.; Beckmann, M.P.; Mohler, K.; Maliszewski, C.R.; Fanslow, W.C.; Lynch, D.H. Pharmacokinetic parameters and biodistribution of soluble cytokine receptors. Int. Rev. Exp. Pathol. 1993, 34, 123–135.
- Klotz, U.; Teml, A.; Schwab, M. Clinical pharmacokinetics and use of infliximab. Clin. Pharmacokinet. 2007, 46, 645–660.
- Vena, G.A.; Cassano, N. Drug focus: Adalimumab in the treatment of moderate to severe psoriasis. Biologics 2007, 1, 93–103.
- Malerich, P.; Elston, D.M. Introduction to TNF/pathophysiology of TNF. In TNF-Alpha Inhibitors; Weinberg, J.M., Buchholz, R., Eds.; Birkhäuser: Basel, Switzerland, 2006; pp. 1–8.
- Tsui, J.J.; Huynh, H.Q. Is top-down therapy a more effective alternative to conventional step-up therapy for Crohn’s disease? Ann. Gastroenterol. 2018, 31, 413–424.
- Allen, P.B.; Bonovas, S.; Danese, S.; Peyrin-Biroulet, L. Evolving primary and secondary endpoints in randomized controlled trials leading to approval of biologics and small molecules in IBD: An historical perspective. Expert. Opin. Biol. Ther. 2020, 20, 151–161.
- Colombel, J.F.; D’Haens, G.; Lee, W.J.; Petersson, J.; Panaccione, R. Outcomes and Strategies to Support a Treat-to-target Approach in Inflammatory Bowel Disease: A Systematic Review. J. Crohns Colitis 2020, 14, 254–266.
- D’Haens, G.; Baert, F.; van Assche, G.; Caenepeel, P.; Vergauwe, P.; Tuynman, H.; De Vos, M.; van Deventer, S.; Stitt, L.; Donner, A.; et al. Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn’s disease: An open randomised trial. Lancet 2008, 371, 660–667.
- Cornillie, F. Ten years of infliximab (remicade) in clinical practice: The story from bench to bedside. Eur. J. Pharmacol. 2009, 623 (Suppl. S1), S1–S4.
- Derkx, B.; Taminiau, J.; Radema, S.; Stronkhorst, A.; Wortel, C.; Tytgat, G.; van Deventer, S. Tumour-necrosis-factor antibody treatment in Crohn’s disease. Lancet 1993, 342, 173–174.
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Tamimoto, Y.; Kimoto, Y.; Uchino, A.; To, K.; Harashima, S.; Hatta, N.; Harada, M. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: Comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008, 58, 1248–1257.
- Kaymakcalan, Z.; Sakorafas, P.; Bose, S.; Scesney, S.; Xiong, L.; Hanzatian, D.K.; Salfeld, J.; Sasso, E.H. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 2009, 131, 308–316.
- Targan, S.R.; Hanauer, S.B.; van Deventer, S.J.; Mayer, L.; Present, D.H.; Braakman, T.; DeWoody, K.L.; Schaible, T.F.; Rutgeerts, P.J. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N. Engl. J. Med. 1997, 337, 1029–1035.
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549.
- Colombel, J.F.; Sandborn, W.J.; Reinisch, W.; Mantzaris, G.J.; Kornbluth, A.; Rachmilewitz, D.; Lichtiger, S.; D’Haens, G.; Diamond, R.H.; Broussard, D.L.; et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N. Engl. J. Med. 2010, 362, 1383–1395.
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2005, 353, 2462–2476.
- Asteria, C.R.; Ficari, F.; Bagnoli, S.; Milla, M.; Tonelli, F. Treatment of perianal fistulas in Crohn’s disease by local injection of antibody to TNF-alpha accounts for a favourable clinical response in selected cases: A pilot study. Scand. J. Gastroenterol. 2006, 41, 1064–1072.
- Shin, D.; Kim, Y.; Kim, Y.S.; Körnicke, T.; Fuhr, R. A Randomized, Phase I Pharmacokinetic Study Comparing SB2 and Infliximab Reference Product (Remicade(®®)) in Healthy Subjects. BioDrugs 2015, 29, 381–388.
- Drobne, D.; Kurent, T.; Golob, S.; Svegl, P.; Rajar, P.; Terzic, S.; Kozelj, M.; Novak, G.; Smrekar, N.; Plut, S.; et al. Success and safety of high infliximab trough levels in inflammatory bowel disease. Scand. J. Gastroenterol. 2018, 53, 940–946.
- European Medicines Agency. Infliximab Sheet. Available online: https://www.ema.europa.eu/en/documents/product-information/remicade-epar-product-information_en.pdf (accessed on 27 June 2022).
- Rutgeerts, P.; D’Haens, G.; Targan, S.; Vasiliauskas, E.; Hanauer, S.B.; Present, D.H.; Mayer, L.; Van Hogezand, R.A.; Braakman, T.; DeWoody, K.L.; et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999, 117, 761–769.
- U.S. Food and Drug Administration (FDA). Adalimumab Label (Humira). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125057s0276lbl.pdf (accessed on 27 June 2022).
- Arora, T.; Padaki, R.; Liu, L.; Hamburger, A.E.; Ellison, A.R.; Stevens, S.R.; Louie, J.S.; Kohno, T. Differences in binding and effector functions between classes of TNF antagonists. Cytokine 2009, 45, 124–131.
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239.
- Colombel, J.-F. The CHARM Trial of Adalimumab in Crohn’s Disease. Gastroenterol. Hepatol. 2006, 2, 486–488.
- Rutgeerts, P.; Van Assche, G.; Sandborn, W.J.; Wolf, D.C.; Geboes, K.; Colombel, J.F.; Reinisch, W.; Investigators, E.; Kumar, A.; Lazar, A.; et al. Adalimumab induces and maintains mucosal healing in patients with Crohn’s disease: Data from the EXTEND trial. Gastroenterology 2012, 142, 1102–1111.e2.
- Panaccione, R.; Loftus, E.V., Jr.; Binion, D.; McHugh, K.; Alam, S.; Chen, N.; Guerette, B.; Mulani, P.; Chao, J. Efficacy and safety of adalimumab in Canadian patients with moderate to severe Crohn’s disease: Results of the Adalimumab in Canadian SubjeCts with ModErate to Severe Crohn’s DiseaSe (ACCESS) trial. Can. J. Gastroenterol. 2011, 25, 419–425.
- Lichtiger, S.; Binion, D.G.; Wolf, D.C.; Present, D.H.; Bensimon, A.G.; Wu, E.; Yu, A.P.; Cardoso, A.T.; Chao, J.; Mulani, P.M.; et al. The CHOICE trial: Adalimumab demonstrates safety, fistula healing, improved quality of life and increased work productivity in patients with Crohn’s disease who failed prior infliximab therapy. Aliment. Pharmacol. Ther. 2010, 32, 1228–1239.
- Sandborn, W.; Van Assche, G.; Reinisch, W. Adalimumab in the Treatment of Moderate-to-Severe Ulcerative Colitis: ULTRA 2 Trial Results. Gastroenterol. Hepatol. 2013, 9, 317–320.
- Thorlund, K.; Druyts, E.; Mills, E.J.; Fedorak, R.N.; Marshall, J.K. Adalimumab versus infliximab for the treatment of moderate to severe ulcerative colitis in adult patients naive to anti-TNF therapy: An indirect treatment comparison meta-analysis. J. Crohns Colitis 2014, 8, 571–581.
- Lee, Y.I.; Park, Y.; Park, S.J.; Kim, T.I.; Kim, W.H.; Cheon, J.H. Comparison of Long-Term Outcomes of Infliximab versus Adalimumab Treatment in Biologic-Naive Patients with Ulcerative Colitis. Gut Liver 2021, 15, 232–242.
- Hyland, E.; Mant, T.; Vlachos, P.; Attkins, N.; Ullmann, M.; Roy, S.; Wagner, V. Comparison of the pharmacokinetics, safety, and immunogenicity of MSB11022, a biosimilar of adalimumab, with Humira((R)) in healthy subjects. Br. J. Clin. Pharmacol. 2016, 82, 983–993.
- Hinojosa, J.; Munoz, F.; Martinez-Romero, G.J. Relationship between Serum Adalimumab Levels and Clinical Outcome in the Treatment of Inflammatory Bowel Disease. Dig. Dis. 2019, 37, 444–450.
- European Medicines Agency. Adalimumab Sheet. Available online: https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf (accessed on 27 June 2022).
- Taleban, S.; Colombel, J.F.; Mohler, M.J.; Fain, M.J. Inflammatory bowel disease and the elderly: A review. J. Crohns Colitis 2015, 9, 507–515.
- U.S. Food and Drug Administration (FDA). Infliximab Label (Remicade). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/103772s5359lbl.pdf (accessed on 27 June 2022).
- Berends, S.E.; Strik, A.S.; Löwenberg, M.; D’Haens, G.R.; Mathôt, R.A.A. Clinical Pharmacokinetic and Pharmacodynamic Considerations in the Treatment of Ulcerative Colitis. Clin. Pharmacokinet. 2019, 58, 15–37.
- Papamichael, K.; Cheifetz, A.S.; Melmed, G.Y.; Irving, P.M.; Vande Casteele, N.; Kozuch, P.L.; Raffals, L.E.; Baidoo, L.; Bressler, B.; Devlin, S.M.; et al. Appropriate Therapeutic Drug Monitoring of Biologic Agents for Patients with Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2019, 17, 1655–1668.e1653.
More