AIntracute respiratory distress syndrome (ARDS) is a heterogeneous syndrome historically characterized by the presence of severe hypoxemia, high-permeability pulmonary edema manifesting as diffuse alveolar infiltrate on chest radiograph, and reduced compliance of the integrated respiratory system as a result of widespread compressive atelectasis and fluid-filled alveoli. Coronavirus disease 19 (COVID-19)-associated ARDS (C-ARDS) is a novel etiology caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that may present with distinct clinical features as a result of the viral pathobiology unique to SARS-CoV-2ranial hypertension is a common finding in severe traumatic brain injury, and requires treatment in the intensive care unit with intracranial pressure monitoring and, when possible, the application of multimodal neuromonitoring. A three-tier approach is suggested in current rec-ommendations, with higher tier therapies having more significant side effects. In this entry, we explain the rationale for this approach, we analyze the benefits and risks of each therapeutic mo-dality, and we discuss how to adapt the therapy to the resources available, based on the most re-cent recommendations.
- COVID-19
- acute respiratory distress syndrome
- mechanical ventilation
- brain trauma
- intracranial hypertension
- neuromonitoring
1. Introduction
Severe traumatic brain injury (TBI) is defined as “an alteration in brain function, or other evidence of brain pathology, caused by an external force”, causing a drop in Glasgow Coma Scale (GCS) to £ 8 [1]. A common characteristic of severe TBI is intracranial hypertension (ICH), which ranges, in different cohorts, from 50-80% of the cases and carries the risk of cerebral herniation [2][3][4][5]. An escalating approach has been adopted for the treatment of ICH, [6][7][8][9][10][11][12] (Figure 1) in which, the different treatment modalities are prioritized according to their efficacy and relative risks of their application [6][8][10][12][13][14]. More difficult to control, refractory ICH, will require higher tier therapies that carry the highest risk of complications. A prerequisite for the application of these treatments is considered the admission of the patient to the intensive care unit (ICU), where these interventions can be applied in the safest possible way. Basic support measures applied in the ICU are considered tier zero, while initial treatments targeting the ICH in particular are considered as tier one treatments (Figure 1). Even though these interventions are not free of complications, a significant effect on survival has recently been attributed to treatments beyond this level [15][16]. For this reason, tier two and three therapies require increased caution, clinical experience and warrant special consideration [17].
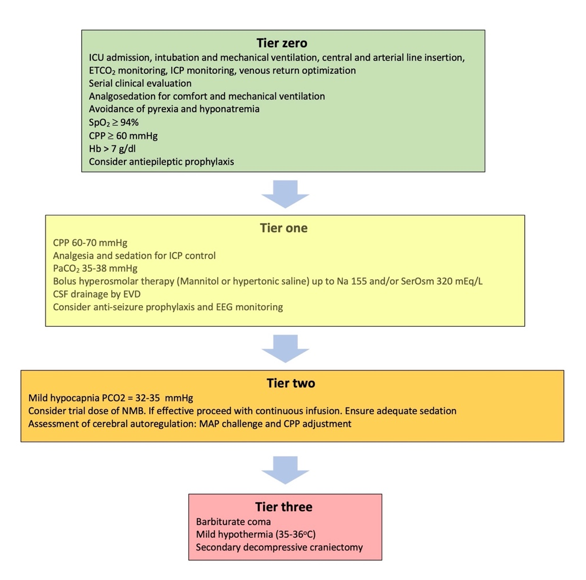
Figure 1. Treatment modalities included in the tiered approach to intracranial hypertension. ICU: Intensive Care unit; ETCO2: End tidal carbon dioxide partial pressure; ICP: Intracranial pressure; SpO2: Oxygen saturation; CPP: Cerebral perfusion pressure; Hb: Hemoglobin concentration; SerOsm: Serum osmolality; CSF: cerebrospinal fluid; EVD: External ventricular drain; EEG: Electroencephalography; NMB: Neuromuscular blocker; MAP: Mean arterial pressure.
2. Tier two therapies
2.1. Mild hypocapnia (PaCO2 32-35 mmHg)
Most clinicians treating patients with TBI know that mild hyperventilation is an effective and rapid way to reduce the ICP by inducing cerebral vasoconstriction and reducing cerebral blood flow [18]. However, it carries the risk of cerebral ischemia [19][20][21][22][23]. One important point is that the basic component for the safe application of hyperventilation is the concomitant application of multimodal neuromonitoring that includes a focal and global assessment of the adequacy of cerebral oxygenation [7][9][24]. In practice, advanced neuromonitoring techniques are not always available in general ICUs, yet, and this limits the range of potentially useful interventions in many TBI patients. In view of such restrictions, and based on current limited evidence, mild hyperventilation is considered as an acceptable measure before escalating to other treatments. Still, close monitoring of the PaCO2 is of paramount importance in order to avoid lowering the PaCO2 to < 30 mmHg [25].
Lowering PCO2 may pose additional problems to trauma patients, besides the risk of brain ischemia [24][26]. It is reasonable to avoid hypocapnia during the first 24 hours after brain trauma, when the blood flow to the brain is known to be reduced [27]. Hypocapnia below 30 mmHg had better be kept as a temporizing measure for the cases of extremes in ICH, when signs of critical neuroworsening (Figure 2) or impending herniation are present. It can then be applied for a limited period of time, as a bridge to higher tier therapies, until other, more appropriate measures are applied. Subsequently, a gradual return to normocapnia is advised, in order to avoid rebound ICH [10].
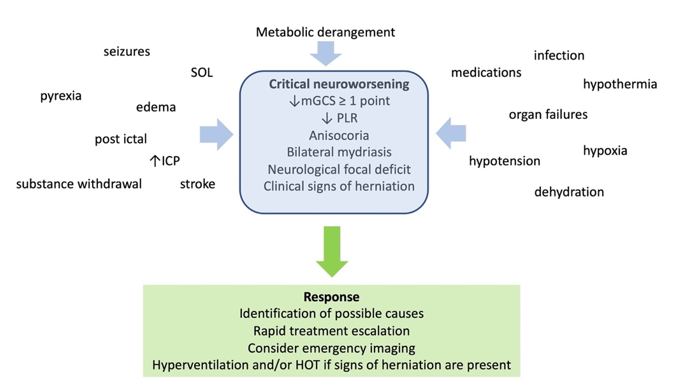
Figure 2. Definition, causes, and management of critical neuroworsening. mGCS: motor Glasgow coma score; PLR: pupillary light reflex; SOL: space occupying lesion; ICP: intracranial pressure; HOT: hyperosmolar therapy
2.2. Neuromuscular blockade (NMB)
The main concerns regarding the use of NMB are the increased risks for ventilator associated pneumonia and its association with ICU - acquired neuromuscular weakness, otherwise termed ICU-neuromyopathy [28][29]. The latter can significantly affect the quality of life of patients discharged from ICUs, and is related to the post intensive care syndrome, which affects more than 60% of ICU survivors [30]. There are trauma patients who also present with lung contusions, acute respiratory distress syndrome (ARDS) or abdominal compartment syndrome, and for whom NMB is otherwise indicated [31][32][33]. The evidence for the effect of NMB on ICH is very limited, with its size reportedly ranging within just 2-3 mmHg. Neuromuscular blocker use is justified during stimulating procedures such as tracheal suction and bronchoscopy, in patients who are deeply sedated. It can also be necessary during the application of cooling measures to lower body temperature, and, since muscular activity significantly contributes to CO2 production, NMB can also assist CO2 control [34]. A trial for NMB is currently suggested for patients in whom ICH is not controlled with tier one measures, with continued infusion reserved for those who show a favorable response [35].
2.3. Assessment of static autoregulation – The mean arterial pressure (MAP) challenge
Cerebral pressure autoregulation can be severely impaired following TBI [36]. While the assessment of dynamic pressure autoregulation requires special equipment, static pressure autoregulation (sPAR) can be evaluated at the patient’s bedside [37][38]. When baseline CPP is above the lower breakpoint of sPAR and then raises further, the resulting vasoconstriction decreases cerebral blood volume, and ICP may also drop. To perform the assessment, the clinician needs to maintain “otherwise stable conditions”, record baseline parameters, titrate vasopressors to a MAP rise of 10 mmHg, and observe and record the response for a maximum of 20 min. Subsequently, MAP/CPP needs to be adjusted, according to whether sPAR is intact or disrupted. The ideal positive response, comprises an ICP drop in response to the MAP rise [36][39].
The assessment of sPAR is associated with several clinical challenges. To perform the test, significant experience is required in treating the increase in ICP caused by the rising MAP when sPAR is disrupted. The adjustment of MAP/CPP is a clinical decision that needs to take into account the relative risks of increasing vasopressor infusion rate. Finally, similarly to dynamic pressure autoregulation status, sPAR is not stable over the clinical course of TBI patients, and may require frequent reassessment [39]. The adjustment of CPP may be particularly challenging in trauma patients who require high vasopressor infusion rates to maintain a MAP of ≥ 70 mmHg (for example), or have concurrent ARDS or cardiac dysfunction [40].
3. Tier three therapies
3.1. Therapeutic hypothermia
Lowering body temperature, and in particular brain temperature, below 36 °C decreases the metabolic demands of brain tissue, hence decreases cerebral blood flow, blood volume and ICP. At the cellular level, hypothermia mitigates calcium induced neurotoxicity, neuronal apoptosis, inflammatory response, and cytotoxic oedema [41]. Despite these experimental findings, these reports did not translate in positive clinical outcomes [42][43][44]. Based on these results, current recommendations suggest the use of mild hypothermia, targeting core body temperatures of 35–36 °C as tier three therapy. Temperatures of < 35 °C are not recommended, due to increased risk for systemic complications [43].
When considering hypothermia, the overall patient’s condition needs to be evaluated in view of the likely side effects. The latter may include impaired cardiac contractility, coagulation and platelet function, increased risk for arrhythmias and infections, and significant fluid and electrolyte shifts [45]. These complications have been reported mainly in patients cooled down to 32–35 °C, while many of them are more pronounced during the rewarming phase [46]. The targeted temperatures can be achieved in many patients with the use of external cooling measures. Cooling blankets or other devices with feedback control should be used when available, in order to avoid lowering body temperature below the desired level, as well as temperature shifts [45]. Compared to other tier three treatments, temperature management may be more suitable for patients without active bleeding and signs of shock, who are not candidates for surgical decompression.
3.2. Metabolic suppression with barbiturates
Barbiturates can induce greater metabolic suppression than midazolam or propofol, and also have antiepileptic properties. By depressing brain function, oxygen consumption and metabolism, they also reduce cerebral blood flow and ICH [6]. They have antiepileptic properties, bind to neuronal γ- aminobutyric acid alpha (GABAA) receptors and cause neuronal hyperpolarization and inhibition of the action potential [47]. In addition, it has been shown that barbiturates reduce lactate and pyruvate production in the brain and inhibit lipid peroxidation mediated by free radicals [48]. These findings suggest that not only are barbiturates effective in the treatment of ICH, but they also possess significant neuroprotective effects. Nevertheless, there is no evidence to support an improvement in clinical outcomes with their use [49].
A plausible argument for this discrepancy is that these sedatives have significant side effects, the most prominent being hemodynamic compromise, hypotension and myocardial depression. Consequently, their use is limited in trauma patients with shock or myocardial injury, since they may further increase vasopressor requirements for the maintenance of adequate CPP [50][51]. Other side effects are immunosuppression, hepatic and renal dysfunction, and suppression of gut motility. At increased doses, barbiturates suppress the pupillary light reflex, in which case patient monitoring relies mainly on invasive measures or imaging. Finally, their use can lead to prolonged sedation due to drug accumulation, and consequently to prolonged need for mechanical ventilation [52][53][54][55]. Dyskalemias, usually appear as hypokalemia during the loading phase and hyperkalemia during withdrawal; the latter may require renal replacement therapy in some cases [56][57].
When metabolic suppression with barbiturates is planned, a test dose should be administered first and the patient’s response should be recorded. A favorable response is characterized by ICP drop and concurrent maintenance of adequate CPP. If this is achieved, then loading doses can be administered. The endpoint of barbiturate administration should be ICP control, and the minimum effective dose should be used in order to minimize the keep side effects. Electroencephalographic monitoring should ideally be applied, in order to titrate the barbiturate infusion rate to a suppression – burst pattern of >> 50%; further increase in barbiturate dose are unlikely to affect ICP [58]. When therapeutic targets are fulfilled and barbiturates are to be withdrawn, a gradual reduction over a few days is advised, so as to avoid hyperkalemia and rebound ICH.
3.3. Decompressive craniectomy
Decompressive craniectomy lowers the ICP, and this has been confirmed by two randomized trials [59][60]. Complications of craniectomy include infections, intracranial hemorrhage, seizures, transcranial herniation, formation of subdural hygroma, and hydrocephalus. These potential complications, along with the risk of cranioplasty [13] and the high baseline risk of a poor neurological outcome, may hinder a decision for secondary decompression. These issues, as well as the probable need for long term care, need to be thoroughly explained to the patients’ families. Decompressive craniectomy remains a tier three therapy, while patients more likely to benefit are those with unilateral pathology, previously fit, with adequate social support, in the setting of adequate medical and social resources. It is also a rescue option in other patients not responding to conservative treatments [13].
4. Conclusions
Tier two and three therapies for ICH in TBI are associated with significant adverse effects and complications. Therefore, these treatments should be chosen when ICH poses a bigger threat to the patient. Current tier concepts allow for flexibility in the choice of therapy and for patient-tailored approaches in different resource settings. Individualized approaches can be achieved by the use of imaging and neuromonitoring modalities, and future research should be oriented toward strengthening the evidence for these treatments and identifying patient profiles that can benefit from each one of them.
References
- Menon DK, Schwab K, Wright DW, Maas AI. Position statement: Definition of traumatic brain injury. Arch Phys Med Rehabil. 2010;91(11):1637-1640. doi:10.1016/j.apmr.2010.05.017
- Vik A, Nag T, Fredriksli OA, et al. Relationship of “dose” of intracranial hypertension to outcome in severe traumatic brain injury. J Neurosurg. 2008;109(4):678-684. doi:10.3171/JNS/2008/109/10/0678
- Kahraman S, Dutton RP, Hu P, et al. Automated measurement of “pressure times time dose” of intracranial hypertension best predicts outcome after severe traumatic brain injury. Journal of Trauma - Injury, Infection and Critical Care. 2010;69(1):110-118. doi:10.1097/TA.0b013e3181c99853
- Güiza F, Depreitere B, Piper I, et al. Visualizing the pressure and time burden of intracranial hypertension in adult and paediatric traumatic brain injury. Intensive Care Med. 2015;41(6):1067-1076. doi:10.1007/s00134-015-3806-1
- Maas AIR, Menon DK, David Adelson PD, et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987-1048. doi:10.1016/S1474-4422(17)30371-X
- Menon DK, Ercole A. Critical care management of traumatic brain injury. In: Handbook of Clinical Neurology. Vol 140. Elsevier B.V.; 2017:239-274. doi:10.1016/B978-0-444-63600-3.00014-3
- Hawryluk GWJ, Aguilera S, Buki A, et al. A management algorithm for patients with intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). In: Intensive Care Medicine. Vol 45. Springer; 2019:1783-1794. doi:10.1007/s00134-019-05805-9
- Stocchetti N, Maas AIR. Traumatic Intracranial Hypertension. New England Journal of Medicine. 2014;370(22):2121-2130. doi:10.1056/nejmra1208708
- Meyfroidt G, Bouzat P, Casaer MP, et al. Management of moderate to severe traumatic brain injury: an update for the intensivist. Intensive Care Med. Published online June 20, 2022. doi:10.1007/s00134-022-06702-4
- Stocchetti N, Carbonara M, Citerio G, et al. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16(6):452-464. doi:10.1016/S1474-4422(17)30118-7
- Stocchetti N, Zoerle T, Carbonara M. Intracranial pressure management in patients with traumatic brain injury: An update. Curr Opin Crit Care. 2017;23(2):110-114. doi:10.1097/MCC.0000000000000393
- Carney N, Totten AM, O’Reilly C, et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery. 2017;80(1):6-15. doi:10.1227/NEU.0000000000001432
- Hawryluk GWJ, Rubiano AM, Totten AM, et al. Guidelines for the management of severe traumatic brain injury: 2020 update of the decompressive craniectomy recommendations. Neurosurgery. 2020;87(3):427-434. doi:10.1093/neuros/nyaa278
- Meyfroidt G, Bouzat P, Casaer MP, et al. Management of moderate to severe traumatic brain injury: an update for the intensivist. Intensive Care Med. 2022;48(6):649-666. doi:10.1007/s00134-022-06702-4
- Huijben JA, Dixit A, Stocchetti N, et al. Use and impact of high intensity treatments in patients with traumatic brain injury across Europe: a CENTER-TBI analysis. Crit Care. 2021;25(1):78. doi:10.1186/s13054-020-03370-y
- Cnossen MC, Huijben JA, van der Jagt M, et al. Variation in monitoring and treatment policies for intracranial hypertension in traumatic brain injury: A survey in 66 neurotrauma centers participating in the CENTER-TBI study. Crit Care. 2017;21(1). doi:10.1186/s13054-017-1816-9
- Gelormini C, Caricato A. “tier-three” therapies in intracranial hypertension: Is it worthwhile? Minerva Anestesiol. 2021;87(12):1287-1289. doi:10.23736/S0375-9393.21.16117-6
- Godoy DA, Seifi A, Garza D, Lubillo-Montenegro S, Murillo-Cabezas F. Hyperventilation therapy for control of posttraumatic intracranial hypertension. Front Neurol. 2017;8(JUL). doi:10.3389/fneur.2017.00250
- Beqiri E, Czosnyka M, Lalou AD, et al. Influence of mild-moderate hypocapnia on intracranial pressure slow waves activity in TBI. Acta Neurochir (Wien). 2020;162(2):345-356. doi:10.1007/s00701-019-04118-6
- Geeraerts T. Moderate hypocapnia for intracranial pressure control after traumatic brain injury: a common practice requiring further investigations. Intensive Care Med. 2021;47(9):1009-1010. doi:10.1007/s00134-021-06489-w
- Godoy DA, Badenes R, Robba C, Murillo Cabezas F. Hyperventilation in Severe Traumatic Brain Injury Has Something Changed in the Last Decade or Uncertainty Continues? A Brief Review. Front Neurol. 2021;12. doi:10.3389/fneur.2021.573237
- Brandi G, Stocchetti N, Pagnamenta A, Stretti F, Steiger P, Klinzing S. Cerebral metabolism is not affected by moderate hyperventilation in patients with traumatic brain injury. Crit Care. 2019;23(1). doi:10.1186/s13054-018-2304-6
- Coles JP, Fryer TD, Coleman MR, et al. Hyperventilation following head injury: Effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35(2):568-578. doi:10.1097/01.CCM.0000254066.37187.88
- Gouvea Bogossian E, Peluso L, Creteur J, Taccone FS. Hyperventilation in Adult TBI Patients: How to Approach It? Front Neurol. 2021;11. doi:10.3389/fneur.2020.580859
- Imberti R, Bellinzona G, Langer M. Cerebral tissue PO2 and SjvO2 changes during moderate hyperventilation in patients with severe traumatic brain injury. J Neurosurg. 2002;96:97-102.
- Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. The Lancet. 2021;398(10300):622-637. doi:10.1016/S0140-6736(21)00439-6
- Cai G, Zhang X, Ou Q, et al. Optimal Targets of the First 24-h Partial Pressure of Carbon Dioxide in Patients with Cerebral Injury: Data from the MIMIC-III and IV Database. Neurocrit Care. 2022;36(2):412-420. doi:10.1007/s12028-021-01312-2
- Murray MJ, Deblock H, Erstad B, et al. Clinical Practice Guidelines for Sustained Neuromuscular Blockade in the Adult Critically Ill Patient. Crit Care Med. 2016;44(11):2079-2103. doi:10.1097/CCM.0000000000002027
- Hermans G, van den Berghe G. Clinical review: Intensive care unit acquired weakness. Crit Care. 2015;19(1). doi:10.1186/s13054-015-0993-7
- Inoue S, Hatakeyama J, Kondo Y, et al. Post‐intensive care syndrome: its pathophysiology, prevention, and future directions. Acute Medicine & Surgery. 2019;6(3):233-246. doi:10.1002/ams2.415
- Papazian L, Forel JM, Gacouin A, et al. Neuromuscular Blockers in Early Acute Respiratory Distress Syndrome. Vol 12.; 2010.
- De laet I, Hoste E, Verholen E, de Waele JJ. The effect of neuromuscular blockers in patients with intra-abdominal hypertension. Intensive Care Med. 2007;33(10):1811-1814. doi:10.1007/s00134-007-0758-0
- Hsiang JK, Chesnut RM, Crisp CB, Klauber MR, Blunt BA, Marshall LF. Early, routine paralysis for intracranial pressure control in severe head injury: is it necessary? Crit Care Med. 1994 Sep;22(9):1471-6. doi: 10.1097/00003246-199409000-00019. PMID: 8062572.
- Mccall M, Jeejeebhoy K, Pencharz P, Moulton R. Effect of Neuromuscular Blockade on Energy Expenditure in Patients With Severe Head Injury. Vol 27.; 2003.
- Sanfilippo F, Santonocito C, Veenith T, Astuto M, Maybauer MO. The Role of Neuromuscular Blockade in Patients with Traumatic Brain Injury: A Systematic Review. Neurocrit Care. 2015;22(2):325-334. doi:10.1007/s12028-014-0061-1
- Rangel-Castilla L, Gasco J, Nauta HJW, Okonkwo DO, Robertson CS. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus. 2008;25(4). doi:10.3171/FOC.2008.25.10.E7
- Donnelly J, Czosnyka M, Adams H, et al. Individualizing Thresholds of Cerebral Perfusion Pressure Using Estimated Limits of Autoregulation. Crit Care Med. 2017;45(9):1464-1471. doi:10.1097/CCM.0000000000002575
- Robba C, Cardim D, Sekhon M, Budohoski K, Czosnyka M. Transcranial Doppler: a stethoscope for the brain-neurocritical care use. J Neurosci Res. 2018;96(4):720-730. doi:10.1002/jnr.24148
- Lang EW, Chesnut RM. A Bedside Method for Investigating the Integrity and Critical Thresholds of Cerebral Pressure Autoregulation in Severe Traumatic Brain Injury Patients. Vol 14.; 2000.
- Annane D, Ouanes-Besbes L, de Backer D, et al. A Global Perspective on Vasoactive Agents in Shock.
- Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: Modulatory effects of hypothermia. J Neuroinflammation. 2010;7. doi:10.1186/1742-2094-7-74
- Zhu Y, Yin H, Zhang R, Ye X, Wei J. Therapeutic hypothermia versus normothermia in adult patients with traumatic brain injury: a meta-analysis. Springerplus. 2016;5(1). doi:10.1186/s40064-016-2391-2
- Hirst TC, Klasen MG, Rhodes JK, MacLeod MR, Andrews PJD. A Systematic Review and Meta-Analysis of Hypothermia in Experimental Traumatic Brain Injury: Why Have Promising Animal Studies Not Been Replicated in Pragmatic Clinical Trials? J Neurotrauma. 2020;37(19):2057-2068. doi:10.1089/neu.2019.6923
- Cooper RJ, Wears RL, Schriger DL. Reporting research results: Recommendations for improving communication. Ann Emerg Med. 2003;41(4):561-564. doi:10.1067/mem.2003.135
- Polderman KH. Application of therapeutic hypothermia in the intensive care unit: Opportunities and pitfalls of a promising treatment modality - Part 2: Practical aspects and side effects. Intensive Care Med. 2004;30(5):757-769. doi:10.1007/s00134-003-2151-y
- Andrews PJD, Sinclair HL, Rodriguez A, et al. Hypothermia for Intracranial Hypertension after Traumatic Brain Injury. New England Journal of Medicine. 2015;373(25):2403-2412. doi:10.1056/NEJMoa1507581
- Zwerus R, Absalom A. Update on anesthetic neuroprotection. Curr Opin Anaesthesiol. 2015;28(4):424-430. doi:10.1097/ACO.0000000000000212
- Almaas R, Saugstad OD, Pleasure D, Rootwelt T. Effect of Barbiturates on Hydroxyl Radicals, Lipid Peroxidation, and Hypoxic Cell Death in Human NT2-N Neurons. Vol 92.; 2000.
- Léger M, Frasca D, Roquilly A, et al. Early use of barbiturates is associated with increased mortality in traumatic brain injury patients from a propensity score-based analysis of a prospective cohort. PLoS One. 2022;17(5):e0268013. doi:10.1371/journal.pone.0268013
- Majdan M, Mauritz W, Wilbacher I, Brazinova A, Rusnak M, Leitgeb J. Barbiturates use and its effects in patients with severe traumatic brain injury in five European countries. J Neurotrauma. 2013;30(1):23-29. doi:10.1089/neu.2012.2554
- Roberts I, Sydenham E. Barbiturates for acute traumatic brain injury. Cochrane Database of Systematic Reviews. Published online December 12, 2012. doi:10.1002/14651858.cd000033.pub2
- Stover JF, Lenzlinger PM, Stocker R, et al. Thiopental in CSF and serum correlates with prolonged loss of cortical activity. Eur Neurol. 1998;39(4):223-228. doi:10.1159/000007938
- Wheeler DW, Thompson AJ, Corletto F, et al. Anaesthetic impairment of immune function is mediated via GABAA receptors. PLoS One. 2011;6(2). doi:10.1371/journal.pone.0017152
- Loop T, Humar M, Pischke S, et al. Thiopental Inhibits Tumor Necrosis Factor-Induced Activation of Nuclear Factor B through Suppression of IB Kinase Activity. Vol 99.; 2003. http://pubs.asahq.org/anesthesiology/article-pdf/99/2/360/408372/0000542-200308000-00017.pdf
- Andrefsky, J.C.; Frank, J.I.; Chyatte, D.; The ciliospinal reflex in pentobarbital coma. J Neurosurg. 1999;90:644-646.
- Cairns CJ, Thomas B, Fletcher S, Parr MJ, Finfer SR. Life-threatening hyperkalaemia following therapeutic barbiturate coma. Intensive Care Med. 2002;28(9):1357-1360. doi:10.1007/s00134-002-1399-y
- Aytuluk HG, Topcu H. Severe hypokalemia and rebound hyperkalemia during barbiturate coma in patients with severe traumatic brain injury. Neurocirugia. 2020;31(5):216-222. doi:10.1016/j.neucir.2019.12.003
- Ellington AL. Electroencephalography and Clinical Neurophysiology CLINICAL AND LABORATORY NOTES ELECTROENCEPHALOGRAPHIC PATTERN OF BURST SUPPRESSION IN A CASE OF BARBITURATE COMA.
- Hutchinson PJ, Kolias AG, Timofeev IS, et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. New England Journal of Medicine. 2016;375(12):1119-1130. doi:10.1056/nejmoa1605215
- Cooper DJ, Rosenfeld J v., Murray L, et al. Decompressive Craniectomy in Diffuse Traumatic Brain Injury. New England Journal of Medicine. 2011;364(16):1493-1502. doi:10.1056/nejmoa1102077.