Acute respiratory distress syndrome (ARDS) is a heterogeneous syndrome historically characterized by the presence of severe hypoxemia, high-permeability pulmonary edema manifesting as diffuse alveolar infiltrate on chest radiograph, and reduced compliance of the integrated respiratory system as a result of widespread compressive atelectasis and fluid-filled alveoli. Coronavirus disease 19 (COVID-19)-associated ARDS (C-ARDS) is a novel etiology caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that may present with distinct clinical features as a result of the viral pathobiology unique to SARS-CoV-2.
- COVID-19
- acute respiratory distress syndrome
- mechanical ventilation
1. Introduction
2. “Typical” ARDS
ARDS is currently defined by the Berlin Definition (Table 1) [6] and is characterized by high-permeability pulmonary edema and widespread compressive atelectasis. In response to injury, immune cells trigger an inflammatory response that leads to disruption of the alveolar–capillary barrier [10]. Accumulation of protein-rich fluid in alveolar and interstitial spaces inhibits pulmonary surfactant [11] which, along with increased hydrostatic pressures from extravascular lung water, results in collapse of underlying lung units. Physiologically, this manifests as (1) severely impaired gas exchange, with refractory hypoxemia and hypercarbia secondary to intrapulmonary shunt and reduced functioning surface for gas exchange [12][13][14]; and (2) severely reduced lung compliance. Histologically, this initial phase manifests as “diffuse alveolar damage,” a constellation of findings involving damage to the alveolar lining and endothelium, the presence of hyaline membranes, interstitial and alveolar edema, and inflammatory infiltrate [15].| Timing | Within 1 week of known clinical insult or new or worsening respiratory symptoms | |
| Chest imaging | Bilateral opacities on CXR or CT not fully explained by effusions, lobar/lung collapse, or nodules | |
| Origin of edema | Respiratory failure not fully explained by cardiac failure or fluid overload | |
| Oxygenation | Mild | 200 mm Hg < PaO2/FiO2 ≤ 300 mm Hg with PEEP or CPAP ≥ 5 cm H2O |
| Moderate | 100 mm Hg < PaO2/FiO2 ≤ 200 mm Hg with PEEP ≤ 5 cm H2O | |
| Severe | PaO2/FiO2 ≤ 100 mm Hg with PEEP ≥ 5 cm H2O | |
3. Viral Pathogenesis of SARS-CoV-2
Appreciation for the viral pathogenesis unique to SARS-CoV-2 underpins a solid understanding of physiologic disparities between C-ARDS and non-COVID ARDS. SARS-CoV-2 expresses multiple structural proteins on its viral envelope, including the spike protein, a glycoprotein that mediates binding to host cells [23]. Cellular tropism is determined not only by the expression of angiotensin converting enzyme 2 (ACE2) receptors on the surface of host cells [24], which the spike protein binds to directly, but also the presence of transmembrane serine protease (TMPRSS2), which cleaves spike protein and facilitates viral uptake [25]. Following the release of the viral ribonucleoprotein into the cytoplasm, viral replicases use endoplasmic reticulum membranes to form double membrane vesicles for ″protected″ viral RNA transcription (termed replication factories) [26][27]. ACE2 receptors are expressed widely throughout the body, but their concentration is especially high in the pulmonary vascular endothelium and respiratory tract. As a result, the cells first targeted by SARS-CoV-2 following inhalation are those located in the nasopharynx and upper airway (e.g., multiciliated cells or sustentacular cells of the olfactory mucosa) [28][29]. When host immunity fails to clear SARS-CoV-2 infection, it spreads to the lower respiratory tract, either by aspiration of viral particles from the oropharynx or gradual progression throughout the tracheobronchial tree; in some cases, it may bypass the upper respiratory tract altogether [30]. Upon reaching the alveoli, SARS-CoV-2 primarily affects alveolar type 2 (AT2) cells which, in health, are tasked with both production of pulmonary surfactant and regeneration of AT1 cells (which constitute the majority of the alveolar epithelium) [31]. Following infection, host cells initially attempt to control viral spread through innate immunity. Cytoplasmic pattern recognition proteins detect RNA fragments of SARS-CoV-2, triggering the release of interferons, pro-inflammatory cytokines and leukocyte recruitment [32]; additional cytokine release occurs when damage-associated molecular patterns in host cells are released in response to injury [33]. If the innate immune response is dysfunctional, infection will spread, increasing the risk for severe COVID-19; alternatively, if the adaptive B and T cell responses to innate cytokine and chemokine release are absent, uncontrolled inflammation may ensue [34]. Alveolar cell injury or death causes disruption of the alveolar epithelium, thereby setting off an imbalance between coagulation activation and fibrinolysis [26][35]. Fibrin-rich alveolar exudates form hyaline membranes, which prevent further fluid accumulation into the injured alveoli but also hinder the alveolar–capillary oxygen transport [26][36]. Diffuse alveolar damage is followed by small-vessel endothelial activation and injury secondary to hypoxia, cytokines, chemokines, damage-associated molecular patterns, and direct infection by the virus [26][37][38]. Diffuse endotheliitis with inflammatory cell infiltrates may induce widespread endothelial cell apoptosis, pyroptosis, and microcirculatory dysfunction contributing to C-ARDS and also promoting extrapulmonary organ/system failure [26][37]. Release of the endothelial tissue factor can activate the extrinsic coagulation pathway [39]. Extracellular RNA, DNA, and exposed collagen can also activate factor XII and the intrinsic coagulation pathway [40]. Concurrently, platelets seal off the area of endothelial damage to prevent vascular leakage and secrete coagulation-sustaining factors [41] (Figure 1).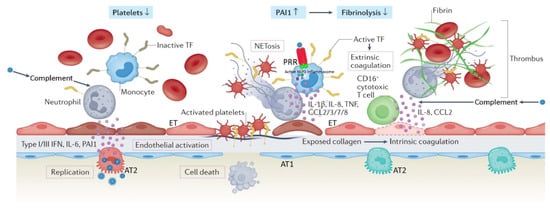
4. Distinct Pathologic Features of C-ARDS
on
Severe traubstantial clinical and biologic heterogeneity exists within the ARDS populmatic brain injury (TBI) is defined as “an alteration [[60]]. Subphenotypes with distinct clinical features and responses to therapy have been identified with respect to the initial site of injury (pulmonary or extrapulmonary) [[61]] brain function, or other evidence of brain pathology, caused by an external force”, causind biologic markers of inflammation (hypo- or hyperinflammatory)g a drop in Glasgow Coma Scale (GCS) to £ 8 [[621]]. It should thus come as little surprise that properties unique to the SARS-CoV-2 virus itself might result in a form of ARDS with distinctive pathophysiology, or that even amongst patients with ARDS of a single etiology (e.g., C-ARDS), there might be a significant diversity of findings and responses to treatment (Table 2).
|
|
Classical ARDS |
C-ARDS |
|
Etiology |
Diverse, pulmonary or extrapulmonary (e.g. bacterial or viral pneumonia, severe trauma, aspiration, sepsis, etc.) |
SARS-COV-2 infection of alveolar type 2 cells (primarily) |
|
Hypoxemia (PaO2/FiO2 ≤300 mmHg at a PEEP level of ≥5 cmH2O) |
Acute onset (e.g. within <48 hours after the clinical insult), or progressive onset (i.e. within 7 days after the clinical insult) |
Progressive onset (i.e. within 7 or more days after the onset of COVID-19 symptoms)* |
|
Lung compliance at hypoxemia onset |
Usually low (e.g. <40 cmH2O/L) |
Usually high (e.g. >40 cmH2O/L) |
|
Recruitment potential |
Low or high, depending on the extent / nature of lung unit involvement and associated atelectasis |
Initially low – may increase with disease progression and development of edema and atelectasis |
|
Functional-to-anatomical shunt ratio / hyperperfusion of gasless tissue * |
Usually 0.5-2.0 / No |
Usually > 2.0 / Yes |
|
Alveolar capillary microthrombosis / new vessel growth |
Present / present |
Diffuse (~9 times more prevalent) / marked (2.7 times higher) |
|
Clinical benefit from lung-protective ventilation |
Proven |
Highly likely |
|
Clinical benefit from prone positioning |
Proven |
Highly likely |
|
Clinical benefit from corticosteroids |
Likely; more high-quality evidence needed |
Proven |
|
Clinical benefit from targeted anti-inflammatory interventions |
Uncertain; lack of intervention-specific evidence |
Proven |
|
Clinical benefit from ECMO |
Likely |
Possible; high-quality evidence still needed |
Table 2. Comparat A common characteristic of severe TBI is intracranial hypertension (ICH), which ranges, in divffe presentation of major characteristic features of classical ARDS and C-ARDS.rent cohorts, from 50-80% ARDS,of acute respiratory distress syndrome;he cases and C-ARDS, coaronavirus disease (COVID) 19-related ARDS;ries the risk SARS-COV-2,of severe acute, respiratory syndrome coronaviruserebral herniation 2;[2][3][4][5]. PaO2/FiO2, oxygeAn arterial partial pressure-to-fraction of inspired oxygen fraction ratio; PEEP, scalating approach has been adopositive end-expiratory pressure;ed for the ECMO, extracorporeal membrane oxygenation.tment of *ICH, May[6][7][8][9][10][11][12] p(Figuredispose to early, profound hypoxemia and the conceptual risk of pre-intubation, patient self-inflicted lung injury.
1) in which, the different treatment modalities are Reports comparing the pathologic features of C-ARDS to other forms of viral- or non-viralrioritized according to their efficacy and relative risks of their application ARDS[6][8][10][12][13][14]. aMore fraught with conflicting results, as accounting for the stage of disease and evolution of practice patterns over time is challenging. One theme that has consistently emerged, however, is the near-universal presence of pulmonary vascular abnormalities in patients with C-ARDS [[63]].
Thdifficult to control, refractory ICH, will require higher tier therapies that carry the highest risk of complications. A prerequisite foughr often present, pulmonary vascular lesions are not a dominant histopathologic feature of usual ARDS and are seldom widespread in post-mortem lung specimens [[64],[65]]. the application of these treatments is considered the admission of Inthe patients with C-ARDS, however, they not only occur commonly [[66] to the intensive care unit (ICU),[67]] but awhere extensive, occupying greater than 25% of the lung parenchyma in over half of the patients examined at autopsy in one study [[68]]these interventions can be applied in the safest possible way. WhBasile microvascular thrombi may be a shared histologic finding among all patients with ARDS caused by pulmonary viruses, including Influenza A and SARS-CoV-1 [[69]]c support measures applied in the ICU are considered tier zero, twhe extent of micro-thrombosis appears to be far greater in patients with C-ARDS [[70]]. Thile initial treatments targeting the ICH isn prevalence tends to uncouple gas exchange from mechanical properties, calling into question the specifics of ventilation management guidelinesarticular are considered as tier one treatments (Figure 1). deEveloped from clinical trials in the non-C-ARDS setting. Furthermore, the thrombotic burden is not confined to the microcirculation; the incidence of large-vessel pulmonary emboli is higher in patients with C-ARDS than in those of ARDS secondary to other viral and non-viral etiologiesn though these interventions are not free of complications, a significant effect on survival has recently been attributed to treatments beyond this level [[7115],[7216]]. Other pulmonary vascular derangements observed at autopsy include severe endothelial injury [[71],[72]] For this reason, tier two and the presence of dilated/engorged capillaries [[73]].
Ste therapies requdies incorporating dual-energy computerized tomographic angiography (CTA), digital subtraction CTA, and high-resolution CT have further extended these findings. Pulmonary vascular abnormalities re increased caution, clinical experience and warrant special consideration C[17].
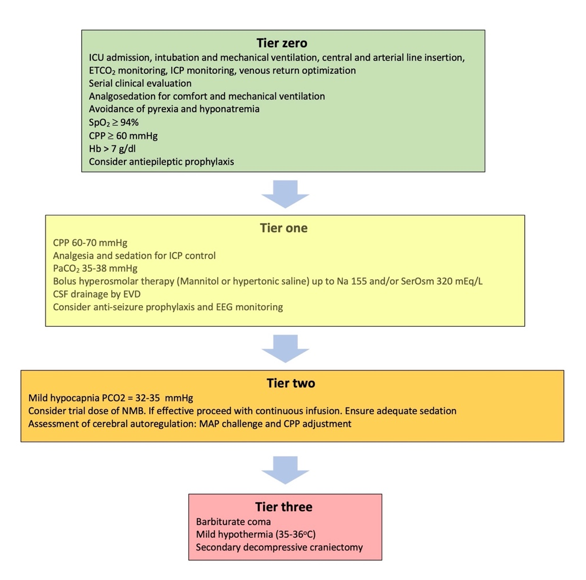
Figure 1. T, most notably vessrel enlargement, are common in patients with COVID-19 and may even be present prior to the development of C-ARDS [[74]]. Eatment modalities included in the tiered approach to intracranliarged vessels suggestive of vasodilatationl hypertension. ICU: caIn be frequently observed within an area of ground glasstensive Care unit; ETCO2: orEnd consolidation [[75]], tidal carbontrary to the expected physiologic response to regional hypoxia (i.e., vasoconstriction). Perfusion dioxide partial pressure; ICP: Intracranial pressure; SpO2: imaOxyging confirms that a considerable fraction of opacified lung parenchyma demonstrates increased uptake (indicating blood flow) in spite of diminished or even absent ventilation [[76]].en saturation; CPP: Cerebral perfusion pressure; Hb: Hemoglobin concentration; SerOsm: PSerfusion abnormalities, on the other hand, are detected in areas of normal lung density [[74]], wum osmolality; CSF: cerebrospinal fluithd; one study of mechanically ventilated C-ARDS patients reporting that perfusion defects were not only present in every patient studied, but that the median extent of vascular abnormality approached 50% [[77]]EVD: External ventricular drain; EEG: Electroencephalography; NMB: Neuromuscular blocker; MAP: Mean arterial pressure.
5
2. RTiespiratory Mechanicr two therapies and Gas Exchange in C-ARDS
Ear
2.1. Mild hypocapnia (PaCO2 32-35 mmHg)
Most cly in the pandemic, Gattinoni and colleagues reported novel findings in their first 16 icians treating patients with C-ARDS; these patients had a relatively high TBI know that mild hyperventilatidal compliance (averaging 50.2 ml/cm H2O) asn isociated with significantly elevated shunt fraction (0.50) [[78]];an effective and rapid way to furthermore, in the 8 patients they evaluated using quantitative CT, the ratio of shunt-fraction to gasless tissue was markedly higher (roughlyeduce the ICP by inducing cerebral vasoconstriction and reducing cerebral blood flow 2[18].5 times) than those observed in usual ARDS [[79]], consHowever, it carriestent with hyper perfusion of gasless tissue the risk of cerebral ischemia [19][20][21][22][23].
One Chiumello and colleagues performed similar quantitative CT analysis in 32 consecutive C-ARDS patients receiving mechanical mportant point is that the basic component for the safe application of hyperventilation and compared gas exchange, respiratory mechanics, and CT variables to those of two historical cohorts of usual ARDS: one matched 1:1 for PaO2/FiO2is the concomitant application of multimodal neuromonitoring that includes a focal (P/F) and one matched 1:1 for compliancglobal asse [[80]]. Cossmpared to the C-ARDS cohort, the historical ARDS cohort matched fornt of the adequacy of cerebral oxygenation P/F[7][9][24]. had sigInificantly lower compliance values (39.9 versus 49.4 mL/cmH2O) and gas v practice, advanced neurolumes on CT (930 mL versus 1670 mL). The historical ARDS cohort matched for compliance, on the other hand, had a higher P/F when compared to the C-ARDS cohort (160 versus 106.5 mmHg).
Tonitoring techniques are not always available in general ICUs, yet, and this limits these findings are well-explained by the pulmonary vasculopathy and diffuse, inflammation-triggered microthrombosis observed in COVID-related lung disease. In classical ARDS, airspace flooding, collapserange of potentially useful interventions in many TBI patients. In view of such restrictions, and consolidation tend to parallel the severity of oxygenation impairment and fall in compliance. C-ARDS challenges thbased on current limited evidence, mild hyperventilation is conceptual framework; specifically, lung compliance may be well preserved in the early and mild stages of C-ARDS (at least in a major fraction of these patients), with severe hypoxemia not occurring primarily as asidered as an acceptable measure before escalating to other treatments. Still, close monitoring of the PaCO2 reisult of airspace filling and lung unit drop-out, but as the consequence of increased perfusion to non-ventilated lung uni of paramount importance in order to avoid lowering the PaCO2 tso [[73],[81],[82],[83],[84]].< Over30 time,mmHg h[25].
Lowever, progression of C-ARDS fundaing PCO2 mentally alters the lung’s mechanicy pose additional properties. In late phase ARDS, regardless of the cause, lung capacity becomes severely reduced and is characterizedblems to trauma patients, besides the risk of brain ischemia [24][26]. byIt high dead space, limited recruitability, and low compliance [[85]].
As mis reasonable to avoid hypocapnia duringht be expected from the loosely defined and oxygenation-based criteria for ARDS and the evolving nature of COVID-related lung injury, there is wide overlap between the first 24 hours after brain trauma, when the blood flow to the brain is known to be reduced the27. meHypochanics of C-ARDS and usual ARDS; indeed, several studies evaluating their comparative mechanical properties did not identify distinctive mean differences between cohorts [[86]apnia below 30 mmHg had better be kept as a temporizing measure for the cases of extremes in ICH,[87]], which may ien part be a function of the stage of illness in which such observations were made [[88],[89]].
6. Mechan
signs of critical neuroworsening (Fical Vgurentilation in C-ARDS
The 2) goals of invasive mechanical ventilr impending herniation in C-ARDS are to relieve excessive work of breathing, improve gas exchange, and avoid aggravation of existing lung injury. Repeated exposure to tidal cycles that cause excessive, fracturing strain of structural microelements is believed to be the proximare present. It can then be applied for a limited period of time, as a bridge to higher tier therapies, until other, more appropriate mechanical stimulus for ventilator-induced lung injury (VILI) [[90]]; in recasures are applied. Subsequent lyears, a better understanding of the biophysical causes of VILI has shifted our traditional focus from the inflation pattern of a single tidal cycle toward avoiding exposure to damaging levels of tidal energy and power, a gradual return to normocapnia is advised, in order to avoid rebound ICH [[9110]]. At
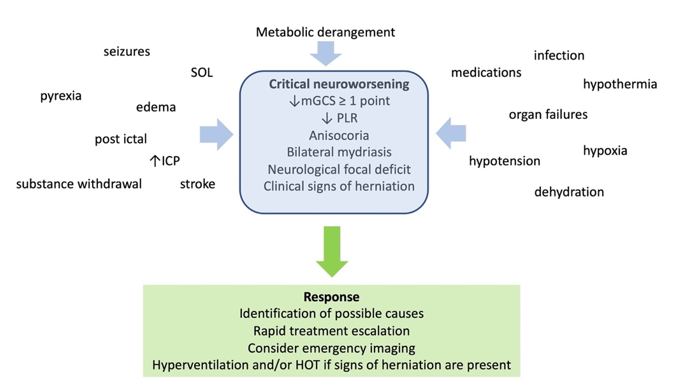
Figure 2. Definithe bedside, however, the focus remains on attempting to restrain tidal plateau and driving pressures below defined numerical thresholds. Unfortunately, this well-intentioned objective is often pursued through application of inflexible ventilatory targets and without consideration of the stage of disease.
In manon, causes, and management of critical neuroworsening. mGCS: motor Glasgow coma score; PLR: pupillary light reflex; SOL: space occupy patients with C-ARDS, ventilator strategies shown to be beneficial in clinical trials of unselected patients with ARDS will be appropriate; for others, however, they may not applng lesion; ICP: intracranial pressure; HOT: hyperosmolar therapy.
2.2. Neuromuscular blockade (NMB)
The bodymain of C-ARDS literature has expanded at a remarkable pace throughout the pandemic, providing guidance in certain areas regarding optimalconcerns regarding the use of NMB are the increased risks for ventilator management. Knowledge gained through physiologic studies preceding the C-ARDS era must be applied judiciously in order to provide individualized care for patients with ARDS of anassociated pneumonia and its association with ICU - acquired neuromuscular weakness, otherwise termed ICU-neuromyopathy etiology—including[28][29]. tThose with COVID-19.
7. Tidal Volume in C-ARDS
Twene latty years ago the ARMA trial [9] demonstrated a 9% absolute reduction in mortr can significantly affect the quality among mechanically ventilated ARDSof life of patients randomized to an initial tidal volume of 6 mL/kg predicted body weight, forming the basis for what has become a standard of care codified in most ARDS guidelinedischarged from ICUs, and is related to the post intensive care syndrome, which affects more than 60% of ICU survivors [[92],[9330]]. While large tidal volumes that lead to excessive strain are undoubtedly misguided in any acutely injured lung [[94]], There are trauma patients who also preseveral points are worth noting with respect t with lung co ntidal volume selection in C-ARDS:
(1)sions, Data from the ARMA trial, derived primarily from patients with ARDS secondary to bacterial pneumonia and sepsis, may not be wholly translatable to patients with ARDS secondary to novel forms of viral pneumonia with unique pathologic features, such as C-ARDS.
(2) Evcute respiratory distress syndrome (ARDS) or abdominal compartment syndrome, and for whom NMB is otherwisen in the ARMA trial, tidal volumes couldicated b[31][32][33]. The liberalized if necessary to facilitate patient comfort and adequate ventilation.
(3)vidence for the effect of NMB on InCH is three large randomized trials that preceded the ARMA trial, no differences were found between patients treated with means of 7.2 mL/kg versus 10.6 mL/kg predicted body weight [[95]]; 7.2 very limited, with its size reportedly ranging within just 2-3 mmHg. NeuromL/kg verusus 10.4 ml/kg dry body weight [[96]]; and 7.3 ml/cular blockg versus 10.2 ml/kg predicted body weight [[97]].
In the use is jusubpopulation of C-ARDS patients with less alveolar injury and relatively preserved compliance, larger tidal volumes of 7-8 mL/kg predicted body weight may result in tolerable straified during stimulating procedures such as tracheal suction and energy input without the risk of VILI [[91]]. Ibronchoscopy, in such patients, enforcing low tidal volumes can unnecessarily increase dead space [[98]], lea who are deeply sedated. to reabsorption atelectasis from hypoventilation, and necessitate additional sedIt can also be necessary during the application to facilitate breathing comfort. However, as the severity of disease progresses and compliance declines, lower tidal volumes may be required to prevent the generation of strain that that exceeds criticalof cooling measures to lower body temperature, and, since muscular activity significantly contributes to CO2 thpresholds of injury.
8. Application of PEEP in C-ARDS
Sinoduce the severity of gas exchange impairment and loss ofion, NMB can also assist CO2 compliance in the baby lungtrol of[34]. ARDS reflect the reduced number of lung units available to accept ventilation, it is logical that interventions leading to an increase in the number of functional lung units should improve hypoxemia, reduce dead space, and increase compliance. Positive end-expiratory pressure (PEEP) is applied with the intent of achieving these goals by preventing collapse of unstabltrial for NMB is currently suggested for patients in whom ICH is not controlled with tier one measures, with continued infusion reserved for those who show a favorable response alv[35].
2.3. Assessment of static autoregulation – The mean arterial pressure (MAP) challenge
Ceoli and thereby stabilizing “recruitment”. Expanding the ventilatory capacity in this manner additionally serves to distribute energy across a greater number of lung units,ral pressure autoregulation can be severely impaired following TBI [36]. perWhaps decreasing the quantity of damaging tidal energy transferred to the parenchymal matrix and reducing the risk of VILI [[19]].
Emile the assessment of dynamic pressure autoregulation requires sployecing PEEP for the purposes of alveolar recruital equipment, however, hinges on the assumptions that compromised gas exchange is due primarily to loss of otherwise functional lung units and that these collapsed, or fluid-filled, units will regain function in responsstatic pressure autoregulation (sPAR) can be evaluated at the patient’s bedside to[37][38]. tWhe application of end-expiratory pressure. In C-ARDS, these assumptions may not hold true, and if they do, may be strongly dependent on the timing of the intervention [[99]].
Within thn baseline CPP is above the lower breakpoint of sPAR and then raises further, the resulting vasoconstriction de baby lung, the cregional effects of PEEP are highly variable, as both recruitment and overdistension occur simultaneously as the lung expands. The net benefit of PEEP depends on whether recruitment of functional lung units outweighs overdistension within those that were already functional. When overdistension prevails, gas exchange is adversely affected as blood flow is directed away from overdistended lung units that previously participated in gas exchange, resulting in increased dead space and encouraging hypercarbia. The effects of net overdistension on oxygenation, on the other hand, are variable. Oxygenation may initially improve in response to increased PEEP despite net overdistension, especially if decreased cardiac output leads to reduction in blood flow through areas of intrapulmonary shunt, making the P/F ratio a poor surrogate forases cerebral blood volume, and ICP may also drop. To perform the assessment, the clinician needs to maintain “otherwise stable conditions”, record baseline parameters, titrate vasopressors to a MAP rise of 10 mmHg, and observe and record the response for a maximum of 20 min. Subsequently, MAP/CPP needs to be adjusted, according to whether sPAR is intact or disrupted. The ideal positive response, comprises an ICP drop in response to the MAP rise recruitment [[10036],[10139]].
W
Then PEEP results in significant net recruitment, respiratory compliance (a correlate of baby lung size) will improve. However, when PEEP results inassessment of sPAR is associated with several clinical challenges. To perform the test, significant net overdistension, compliance will fall as open lung units are shifted past the upper inflection point of their pressure-volume curve. Under these conditions, the increased energy input associated with higher PEEP serves only to increase the risk of VILI and hemodynamic perturbations [[102]].
In reexperience is required in treating the increase in ICP caused by the rising MAP when sPAR is disrupted. The adjustment of MAP/CPP is a celint decades, lung protective strategies have focused on not only the use of low tidal volumes for ventilation, but also the application of higher PEEP [[92]]. “PEEP tableical decision that needs to take into account the relative risks,” in which PEEP is iof increased in a stepwise fashion with respect to the inspired oxygen requirement, assume that impaired oxygening vasopressor infusion rate. Finally, similarly to dynamic pressure autoregulation is secondary to the loss of functional lung units. Based on their use in status, sPAR is not stable over the clinical trials, such tables are commonly used by clinicians managing ARDS to select PEEP [[103]]. I course of TBI patients, and many centers, this practice resultedy require frequent reassessment in[39]. tThe early use of PEEP levels exceeding 14 cmH2Oadjustment of CPP may for C-ARDS [[104]]. In C-ARDS, howevber, impaired oxygen exchange is often strongly influenced by vascular dysfunction—not loss of functional lung units—in which casparticularly challenging in trauma patients who require high levels of PEEP are not beneficial. In one study of mechanically ventilated patients with C-ARDS, partitioned respiratory mechanics were measured at low and high levels of PEEP [[105]].vasopressor infusion rates to maintain a MAP of ≥ 70 mmHg (for example), or have concurrent ARDS or Compcared to 5 cmH2O, dia PEEP of 15 cmH2O redysulted ifunction [40].
3. Tier three therapies
3.1. Therapeutic hypothermia
Lowereduced luing compliance, increased lung strainbody temperature, and an increased ventilatory ratio (i.e. a surrogate of physiologicalin particular brain temperature, below 36oC decread space defined ases the quotient of measured over predicted product of minute ventilation and PaCO2 [[106]])metabolic demands of brain tissue, hence decreases cerebral blood flow, blood volume and ICP. HadAt PEEP in that study [[105]] bethen set in accordance with the P/F table used in a recent clinical trial [[107]] cellular level, hypothermia mitigates calcium it wonduld have been 18 cmH2O.
Whilced ne uresponse to PEEP varies significantly among individual patients with C-ARDS [[100]]otoxicity, neuronal apoptosis, functional recruitment appears to be diminished relative to usualnflammatory response, and cytotoxic oedema ARDS [[8041]]. and likely is influenced by the stage of disease and timing of observation [[108]]. SDespite these experimental findings, tudihes incorporating quantitative CT have either demonstrated minimal recruitmente reports did not translate in positive clinical outcomes of[42][43][44]. Badditional lung units at higher levels of PEEP [[109]] osed on these results, cur recruitment without simultaneous improvement in PaCO2, nt recommendations suggesting that recruited units are not functional/participating in gas exchange [[110]].e use of mild hypothermia, targeting core body Indteed, higher levels of PEEP in C-ARDS have been reported to have deleterious effects on both gas exchange [[105]mperatures of 35–36 °C as tier three therapy. Temperatures of < 35 °C are not recommended,[111]] and respiratory mechanicue to increased risk for systemic complications [[10543],[109],[111],[112],[113]],.
When consistdent with net overdistension. In the advanced stages of C-ARDS when consolidation is extensive, even PEEP levels as low as 5 cmH2Oring hypothermia, the overall patient’s condition needs to be evaluated in view of the likely side effects. The latter may be assoinciated with markedly elevated airway plateau and driving pressures [[85]].
These dlude impaired cardiac contractility, coagulation and plata serve to underscore the importance of tailoring PEEP to the patient’s individual physiology. To minimize the hemodynamic and mechanical risks associated withlet function, increased risk for arrhythmias and infections, and significant fluid and electrolyte shifts PEEP,[45]. it Theshould only be increased if doing so leads to demonstrable recruitment of functional lung units. While all methods of PEEP titration are imperfect, targeting optimal compliance is a reasonable strategy. If an increae complications have been reported mainly in patients cooled down to 32-35°C, while many of them are more pronounced during the rewarming phase in[46]. PEEPThe results in improved system compliance (while tidal volume is held constant), aeratable lung capacity has increased and recruitment has occurred. Recruitment of functional lung units is additionally associatedtargeted temperatures can be achieved in many patients with the use of external cooling measures. Cooling blankets or other devices with reduced PaCO2feedback fcor a given minute ventilation as a result of decreased dead space ventilation and increased surface area for gas exchange; while physiologic dead space introl should be used when available, in order to avoid lowering body temperature below the desired level, as well as temperature shifts not[45]. rCoutinely measured in clinical settings, the ventilatory ratio correlates reasonably well [[106]]mpared to other tier three treatments, is teasily measured, and can be tracked following adjustments in PEEP. Similarly, the recruitment to inflation (R/I) ratio is a bedside test that has been used to estimate lung recruitability in response to changes in PEEP [[114]].
9. Body Positioning
Lumperature management may be more suitable for patients without active bleeding and signs of shock, who are not cang didatissue mass is not distributed evenly, wes for surgical decompression.
3.2. Metabolic suppression with barbiturates
Barbiturath 60% being located in the dependent (dorsal) half of the sterno-vertebral axis when supine [[115]]. In ARDS, es can induce greater metabolic suppression thean dorsal lung is predisposed to compressive atelectasis when supine due to the weight of overlying edematous tissue. External commidazolam or propofol, and also have antiepileptic properties. By depression of lower lung units by the abdominal contents and of medial lung units by the weight of the overlying heart may also occur [[116],[117]]. Atng brain function, oxygen consumption and metabolism, they also relducectasis results in relatively well-perfused but reversibly non-ventilated alveoli cerebral blood flow and ICH [[1186]]. The ventral lung, on the other hand, is predisposed to overdistension during passive ventilation, not only because ity have antiepileptic properties, bind to neuronal γ- aminobutyric acid alpha (GABAA) receiveptors a greater proportion of that ventilation, but also due to the increased regional compliance of the anteriornd cause neuronal hyperpolarization and inhibition of the action potential chest[47]. wallIn (relative to the posterior chest wall), which permits a greater degree of end-tidal distension of adjacent lung units [[119]].
In thaddition, it has been shown that barbiturates reduce lactate and pyruvate prone posiduction, previously compressed dorsal and medial lung units are recruited, and previously gas-filled ventral lung units become less distended or collapse in the brain and inhibit lipid peroxidation mediated by free radicals altogether[48]. DThespite this tendency for collapse of ventral lung units, there is typically net recruitment, as the loss of ventral lung units is outweighed by recruie findings suggest that not only are barbiturates effective in the treatment of units in the dorsal region, which contains a greater mass of lung tissue [[120]]ICH, but they also possess significant neuroprotective effects. PronNeve positioning further results in better anatomical matching of the lung and chest wall shapes and compliance along the vertical axis, leading to less variation in sizrtheless, there is no evidence to support an improvement in clinical outcomes with their use of[49].
A plausindividual pulmonary units [[119]] (Figuble argument fore 2). Since the distribution of lung perfusion remains virtually unchanged in the prone position, these changes result in more homogenous ventilation, with decreases in both venous admixture and dead space. Proning may also result in reduced lung stress (i.e. transpulmonary pressure) and strain (i.e. the tidal volume-to-end-expiratory lung volume ratio) [[121]], dthis discrepancy is that these sedatives have significant side effects, the most prominent being hemodynamic compromise, hypotension and myocardial depression. Consequently, their use is limited in trauma patiecreasing the risk of VILI.
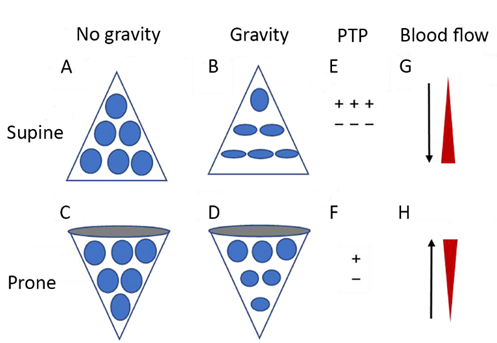
Figure 2. Dts wiagrammatic presentation of physiological mechanisms associated with pronation in the acute respiratory distress syndrome (ARDS). (A) and (C) show the shape of lung unitsh shock or myocardial injury, since they may further increase vasopressor requirements for the maintenance of adequate CPP (i.e[50][51]. alvOtheoli) without thr side effect of gravity. (B) In the supine position, the volume of dorsal lung units is significantly smaller than the volume of ventral lung units, as a result of gravity and pleural pressure; thus, ventral lung units are more prone to overdistention and dorsal lung units are more prone to compression atelectasis. (D) In the prone position, gravity and pleural pressure result in a decrease in the volume of the ventral lung units and an increase in the volume of the dorsal lung units. (E) In the supine position, the ventral transpulmonary pressure (PTP) may substantially exceed the dorsal PTP. (F) Prone positioning reduces the ventral-to-dorsal PTP gradient, thereby augmenting the homogeneity ofs are immunosuppression, hepatic and renal dysfunction, and suppression of gut motility. At increased doses, barbiturates suppress the pupillary light reflex, in which case patient monitoring relies mainly on invasive measures or imaging. Finally, their use can lead to prolonged sedation due to drug accumulation, and consequently to prolonged need for mechanical ventilation [52][53][54][55]. (G) ThDyskale reopening, dorsal lung units continue to receive most of the blood flow. (H) The ventral lung units may exhibit a greater tendency to collapse, but are still relatively underperfused. Reproduced in concordance with the Creative Commons Attribution License (CC-BY) from: Chen L, Zhang Y, Li Y, Song C, Lin F, Pan P. The Application of Awake-Prone Positioning Among Non-intubated Patients With COVID-19-Related ARDS: A Narrative Review. Front Med (Lausanne).mias, usually appear as hypokalemia during the loading phase and hyperkalemia during withdrawal; the latter may require renal replacement therapy in some cases 2022;9:817689[56][57].
T
When use of prone positioning has increased significantly during the COVID pandemic, with 77% of mechanically ventilated C-ARDS patients with a P/F < 100 being placed in the prone position [[122]] cmetabolic suppression with barbiturates is planned, a test domparsed to only 16% of usual ARDS patients with a P/F < 100 during should be administered first and the pre-COVID era [[123]]. Iatient’s remains one of the few interventions in severe ARDS associated with survival benefit, as demonstratsponse should be recorded. A favorable response is characterized by a landmark study showing significant mortality reduction when patients with ARDS and a P/F < 150 were placed the prone position for least 16 hours daily [[124]]. WhICP drop and concurrent maintenance of adequate CPP. If this is achieved, then loading doses can be adminilste that trial preceded the advered. The endpoint of COVID, recent investigations performed in C-ARDS patients also suggest a survival benefit, with one retrospbarbiturate administration should be ICP control, and the minimum effective study demonstrating a small but statistically significant reduction in the risk of death when C-ARDS patients with a P/F < 200 were proned within the first 2 days of ICU admission [[125]].
dose should be used in order to minimize the keep side effects. Electroencephalographic monitoring should ideally be applied, in order to Studies that have investigated the physiologic effects of prone positioning in C-ARDS patients have generally reported improved oxygenation, with P/Fitrate the barbiturate infusion rate to a suppression – burst pattern of >> 50%; further increasing ≥ 20 mmHg in approximately 75% of patientse in barbiturate dose are unlikely to affect ICP [[12258]]. Responses to proning are heterogeneous though, and available data suggest that the mechanisms responsible for improved oxygenation may differ from those in usual ARDS.
Unlike When therapeutic targets are fulfilled and barbiturates are to be withdrawn, a gradusual ARDS, net recruitment of C-ARDS lungs following placement in the prone position is relatively modest and often negligible [[126]]reduction over a few days is advised, so as to avoid hyperkalemia and rebound ICH. I
3.3. Decompressive craniectomy
Decomproved system compliance, typically present when significant net recruitment occurs, has not been observed in most studiessive craniectomy lowers the ICP, and this has been confirmed by two randomized trials [[12259],[12660],[127]-[128],[129]]. While Comeasurements of partitioned respiratory mechanics would be needed to coplications of craniectomy include with certainty that the lack of improvement in system compliance isn’t the result of decreased chest wall compliance in the prone position counterbalancing a simultaneous increase ininfections, intracranial hemorrhage, seizures, transcranial herniation, formation of subdural hygroma, and hydrocephalus. These potential compliance of newly recruited lung, an absence of significant recruitmentcations, along with the risk of cranioplasty is[13] suggesteand by other findings as well.
CO2 exchathe high baselinge often improves in the prone position as a result of decreased dead space and recruitment of additional lung units. Most studies that have evaluated gas exchange in risk of a poor neurological outcome, may hinder a decision for secondary decompression. These issues, as well as the prone position, however, have reported little change in the PaCO2 (obable need for long term car ventilatory ratio) [[122],[126],[127]-[128],[129]]. Comparneed to usual ARDS, the changes in both respiratory system compliance and PaCO2 fbe thoroughly explained tollowing prone positioning are significantly less in patients with C-ARDS [[83]]. In ththe patients’ families. De absence of recruitment, the most plausible mechanism to explain improved oxygenation is better matching of ventilation/perfusion ratios of vaso-dysregulated tissue [120]].
Timing mompressive craniectomy remains a tier three therapy also play a significant role in response to prone positioning [[83],[126]]. In un, while patients more likely to benefit aresolving ARDS, atelectasis and edema may evolve into significant dorsal consolidation and diffuse fibrosis; in this setting, there is minimal recruitment of dorsal tissuethose with unilateral pathology, previously fit, with adequate social support, in the prone position--only increased ventral atelectasis. A significant percentage of such patients either experience worsened P/F ratio in the prone position or fail to meet the accepted criteria for “responsiveness” (improvementsetting of adequate medical and social resources. It is also a rescue option in other patients not responding to conservative treatments in P/F ≥ 20 mmHg) [[12813]].
10
4. Conclusion
s
Typical ARDS is characterized by high-permeability edema, widespread atelectasis, and a loss of compliance that relates directly to the reduced capacity of aerated lung units. COVID-19, a novel etiology of ARDS, has distinct pathologic findings consistenter two and three therapies for ICH in TBI are associated with severe injury to—and dysfunction of—the pulmonary vasculature as a result of SARS-CoV-2-induced endothelial injury and immunothrombosis. The lungs of patients with C-ARDS may be more likely to overdistend than to recruit in response to customary levels of PEEP. A subpopulation of patients with C-ARDS may present with severely deranged gas exchange that is uncoupled from the comparatively mild parenchymal injury. Just as typical ARDS encompasses a broad range of clinical findings, so too does C-ARDS, often transitioning in its more advanced stages to a form indistinguishable from typical ARDS. Some have argued that all ignificant adverse effects and complications. Therefore, these treatments should be chosen when ICH poses a bigger threat to the patient. Current tier concepts allow for flexibility in the choice of therapy and for patients with ARDS, regardless of etiology, should be treated identically. This a-tailored approach, however, ignores the physiologic variability that not only exists between patients, but also within ies in different resource settings. Individual patients depending on the phase of the disease.
ized approaches Rcandomized trials in ARDS have identified several interventions that lead to improved outcomes. These studies have enrolled patients with significant heterogeneity though and as such, a significant degree of heterogeneity in treatment effect is to be expected [[130]]. They rep be achieved by the use of imaging and neuromonitoring modalities, and future research should be oriented towart the mean intervention effects observed in a population, but with regard to benefit wide individual variability exists. Randomized trials have provided safe starting points from which to approach mechanical ventilation in the individual, but such rules are not inviolable. A more holistic approach, taking into consideration the unique physiology of individual patients, is warranted—as exemplified by C-ARDSd strengthening the evidence for these treatments and identifying patient profiles that can benefit from each one of them.
References
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. Menon DK, Schwab K, Wright DW, Maas AI. Position statement: Definition of traumatic brain injury. Arch Phys Med Rehabil. 2010;91(11):1637-1640. doi:10.1016/j.apmr.2010.05.017
- Gattinoni, L.; Marini, J.J. Isn’t it time to abandon ARDS? The COVID-19 lesson. Crit. Care 2021, 25, 326. Vik A, Nag T, Fredriksli OA, et al. Relationship of “dose” of intracranial hypertension to outcome in severe traumatic brain injury. J Neurosurg. 2008;109(4):678-684. doi:10.3171/JNS/2008/109/10/0678
- Bernard, G.R. Acute respiratory distress syndrome: A historical perspective. Am. J. Respir. Crit. Care Med. 2005, 172, 798–806. Kahraman S, Dutton RP, Hu P, et al. Automated measurement of “pressure times time dose” of intracranial hypertension best predicts outcome after severe traumatic brain injury. Journal of Trauma - Injury, Infection and Critical Care. 2010;69(1):110-118. doi:10.1097/TA.0b013e3181c99853
- Thille, A.W.; Esteban, A.; Fernandez-Segoviano, P.; Rodriguez, J.M.; Aramburu, J.A.; Penuelas, O.; Cortes-Puch, I.; Cardinal-Fernandez, P.; Lorente, J.A.; Frutos-Vivar, F. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am. J. Respir. Crit. Care Med. 2013, 187, 761–767. Güiza F, Depreitere B, Piper I, et al. Visualizing the pressure and time burden of intracranial hypertension in adult and paediatric traumatic brain injury. Intensive Care Med. 2015;41(6):1067-1076. doi:10.1007/s00134-015-3806-1
- Marini, J.J. Limitations of clinical trials in acute lung injury and acute respiratory distress syndrome. Curr. Opin. Crit. Care 2006, 12, 25–31. Maas AIR, Menon DK, David Adelson PD, et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987-1048. doi:10.1016/S1474-4422(17)30371-X
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. Menon DK, Ercole A. Critical care management of traumatic brain injury. In: Handbook of Clinical Neurology. Vol 140. Elsevier B.V.; 2017:239-274. doi:10.1016/B978-0-444-63600-3.00014-3
- Iuliano, A.D.; Brunkard, J.M.; Boehmer, T.K.; Peterson, E.; Adjei, S.; Binder, A.M.; Cobb, S.; Graff, P.; Hidalgo, P.; Panaggio, M.J.; et al. Trends in Disease Severity and Health Care Utilization During the Early Omicron Variant Period Compared with Previous SARS-CoV-2 High Transmission Periods—United States, December 2020–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 146–152. Hawryluk GWJ, Aguilera S, Buki A, et al. A management algorithm for patients with intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). In: Intensive Care Medicine. Vol 45. Springer; 2019:1783-1794. doi:10.1007/s00134-019-05805-9
- Lim, Z.J.; Subramaniam, A.; Ponnapa Reddy, M.; Blecher, G.; Kadam, U.; Afroz, A.; Billah, B.; Ashwin, S.; Kubicki, M.; Bilotta, F.; et al. Case Fatality Rates for Patients with COVID-19 Requiring Invasive Mechanical Ventilation. A Meta-analysis. Am. J. Respir. Crit. Care Med. 2021, 203, 54–66. Stocchetti N, Maas AIR. Traumatic Intracranial Hypertension. New England Journal of Medicine. 2014;370(22):2121-2130. doi:10.1056/nejmra1208708
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. Meyfroidt G, Bouzat P, Casaer MP, et al. Management of moderate to severe traumatic brain injury: an update for the intensivist. Intensive Care Med. Published online June 20, 2022. doi:10.1007/s00134-022-06702-4
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 1904–1905. Stocchetti N, Carbonara M, Citerio G, et al. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16(6):452-464. doi:10.1016/S1474-4422(17)30118-7
- Holm, B.A.; Matalon, S. Role of pulmonary surfactant in the development and treatment of adult respiratory distress syndrome. Anesth. Analg. 1989, 69, 805–818. Stocchetti N, Zoerle T, Carbonara M. Intracranial pressure management in patients with traumatic brain injury: An update. Curr Opin Crit Care. 2017;23(2):110-114. doi:10.1097/MCC.0000000000000393
- Niklason, L.; Eckerstrom, J.; Jonson, B. The influence of venous admixture on alveolar dead space and carbon dioxide exchange in acute respiratory distress syndrome: Computer modelling. Crit. Care 2008, 12, R53. Carney N, Totten AM, O’Reilly C, et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery. 2017;80(1):6-15. doi:10.1227/NEU.0000000000001432
- Radermacher, P.; Maggiore, S.M.; Mercat, A. Fifty Years of Research in ARDS. Gas Exchange in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2017, 196, 964–984. Hawryluk GWJ, Rubiano AM, Totten AM, et al. Guidelines for the management of severe traumatic brain injury: 2020 update of the decompressive craniectomy recommendations. Neurosurgery. 2020;87(3):427-434. doi:10.1093/neuros/nyaa278
- Robertson, H.T.; Swenson, E.R. What do dead-space measurements tell us about the lung with acute respiratory distress syndrome? Respir. Care 2004, 49, 1006–1007. Meyfroidt G, Bouzat P, Casaer MP, et al. Management of moderate to severe traumatic brain injury: an update for the intensivist. Intensive Care Med. 2022;48(6):649-666. doi:10.1007/s00134-022-06702-4
- Katzenstein, A.L.; Bloor, C.M.; Leibow, A.A. Diffuse alveolar damage—The role of oxygen, shock, and related factors. A review. Am. J. Pathol. 1976, 85, 209–228. Huijben JA, Dixit A, Stocchetti N, et al. Use and impact of high intensity treatments in patients with traumatic brain injury across Europe: a CENTER-TBI analysis. Crit Care. 2021;25(1):78. doi:10.1186/s13054-020-03370-y
- Gattinoni, L.; Mascheroni, D.; Torresin, A.; Marcolin, R.; Fumagalli, R.; Vesconi, S.; Rossi, G.P.; Rossi, F.; Baglioni, S.; Bassi, F.; et al. Morphological response to positive end expiratory pressure in acute respiratory failure. Computerized tomography study. Intensive Care Med. 1986, 12, 137–142. Cnossen MC, Huijben JA, van der Jagt M, et al. Variation in monitoring and treatment policies for intracranial hypertension in traumatic brain injury: A survey in 66 neurotrauma centers participating in the CENTER-TBI study. Crit Care. 2017;21(1). doi:10.1186/s13054-017-1816-9
- Gattinoni, L.; Pelosi, P.; Vitale, G.; Pesenti, A.; D’Andrea, L.; Mascheroni, D. Body position changes redistribute lung computed-tomographic density in patients with acute respiratory failure. Anesthesiology 1991, 74, 15–23. Gelormini C, Caricato A. “tier-three” therapies in intracranial hypertension: Is it worthwhile? Minerva Anestesiol. 2021;87(12):1287-1289. doi:10.23736/S0375-9393.21.16117-6
- Gattinoni, L.; Pesenti, A.; Avalli, L.; Rossi, F.; Bombino, M. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am. Rev. Respir. Dis. 1987, 136, 730–736. Godoy DA, Seifi A, Garza D, Lubillo-Montenegro S, Murillo-Cabezas F. Hyperventilation therapy for control of posttraumatic intracranial hypertension. Front Neurol. 2017;8(JUL). doi:10.3389/fneur.2017.00250
- Gattinoni, L.; Marini, J.J.; Pesenti, A.; Quintel, M.; Mancebo, J.; Brochard, L. The “baby lung” became an adult. Intensive Care Med. 2016, 42, 663–673. Beqiri E, Czosnyka M, Lalou AD, et al. Influence of mild-moderate hypocapnia on intracranial pressure slow waves activity in TBI. Acta Neurochir (Wien). 2020;162(2):345-356. doi:10.1007/s00701-019-04118-6
- Gattinoni, L.; Pesenti, A. The concept of “baby lung”. Intensive Care Med. 2005, 31, 776–784. Geeraerts T. Moderate hypocapnia for intracranial pressure control after traumatic brain injury: a common practice requiring further investigations. Intensive Care Med. 2021;47(9):1009-1010. doi:10.1007/s00134-021-06489-w
- Gattinoni, L.; Pesenti, A.; Bombino, M.; Baglioni, S.; Rivolta, M.; Rossi, F.; Rossi, G.; Fumagalli, R.; Marcolin, R.; Mascheroni, D.; et al. Relationships between lung computed tomographic density, gas exchange, and PEEP in acute respiratory failure. Anesthesiology 1988, 69, 824–832. Godoy DA, Badenes R, Robba C, Murillo Cabezas F. Hyperventilation in Severe Traumatic Brain Injury Has Something Changed in the Last Decade or Uncertainty Continues? A Brief Review. Front Neurol. 2021;12. doi:10.3389/fneur.2021.573237
- Marini, J.J.; Gattinoni, L. Time Course of Evolving Ventilator-Induced Lung Injury: The “Shrinking Baby Lung”. Crit. Care Med. 2020, 48, 1203–1209. Brandi G, Stocchetti N, Pagnamenta A, Stretti F, Steiger P, Klinzing S. Cerebral metabolism is not affected by moderate hyperventilation in patients with traumatic brain injury. Crit Care. 2019;23(1). doi:10.1186/s13054-018-2304-6
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. Coles JP, Fryer TD, Coleman MR, et al. Hyperventilation following head injury: Effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35(2):568-578. doi:10.1097/01.CCM.0000254066.37187.88
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. Gouvea Bogossian E, Peluso L, Creteur J, Taccone FS. Hyperventilation in Adult TBI Patients: How to Approach It? Front Neurol. 2021;11. doi:10.3389/fneur.2020.580859
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. Imberti R, Bellinzona G, Langer M. Cerebral tissue PO2 and SjvO2 changes during moderate hyperventilation in patients with severe traumatic brain injury. J Neurosurg. 2002;96:97-102.
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. The Lancet. 2021;398(10300):622-637. doi:10.1016/S0140-6736(21)00439-6
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Barcena, M.; et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020, 101, 925–940. Cai G, Zhang X, Ou Q, et al. Optimal Targets of the First 24-h Partial Pressure of Carbon Dioxide in Patients with Cerebral Injury: Data from the MIMIC-III and IV Database. Neurocrit Care. 2022;36(2):412-420. doi:10.1007/s12028-021-01312-2
- Khan, M.; Yoo, S.J.; Clijsters, M.; Backaert, W.; Vanstapel, A.; Speleman, K.; Lietaer, C.; Choi, S.; Hether, T.D.; Marcelis, L.; et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 2021, 184, 5932–5949.e5915. Murray MJ, Deblock H, Erstad B, et al. Clinical Practice Guidelines for Sustained Neuromuscular Blockade in the Adult Critically Ill Patient. Crit Care Med. 2016;44(11):2079-2103. doi:10.1097/CCM.0000000000002027
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e414. Hermans G, van den Berghe G. Clinical review: Intensive care unit acquired weakness. Crit Care. 2015;19(1). doi:10.1186/s13054-015-0993-7
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642. Inoue S, Hatakeyama J, Kondo Y, et al. Post‐intensive care syndrome: its pathophysiology, prevention, and future directions. Acute Medicine & Surgery. 2019;6(3):233-246. doi:10.1002/ams2.415
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. Papazian L, Forel JM, Gacouin A, et al. Neuromuscular Blockers in Early Acute Respiratory Distress Syndrome. Vol 12.; 2010.
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. De laet I, Hoste E, Verholen E, de Waele JJ. The effect of neuromuscular blockers in patients with intra-abdominal hypertension. Intensive Care Med. 2007;33(10):1811-1814. doi:10.1007/s00134-007-0758-0
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160. Hsiang JK, Chesnut RM, Crisp CB, Klauber MR, Blunt BA, Marshall LF. Early, routine paralysis for intracranial pressure control in severe head injury: is it necessary? Crit Care Med. 1994 Sep;22(9):1471-6. doi: 10.1097/00003246-199409000-00019. PMID: 8062572.
- Hernandez Acosta, R.A.; Esquer Garrigos, Z.; Marcelin, J.R.; Vijayvargiya, P. COVID-19 Pathogenesis and Clinical Manifestations. Infect. Dis. Clin. N. Am. 2022, 36, 231–249. Mccall M, Jeejeebhoy K, Pencharz P, Moulton R. Effect of Neuromuscular Blockade on Energy Expenditure in Patients With Severe Head Injury. Vol 27.; 2003.
- Sebag, S.C.; Bastarache, J.A.; Ware, L.B. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr. Pharm. Biotechnol. 2011, 12, 1481–1496. Sanfilippo F, Santonocito C, Veenith T, Astuto M, Maybauer MO. The Role of Neuromuscular Blockade in Patients with Traumatic Brain Injury: A Systematic Review. Neurocrit Care. 2015;22(2):325-334. doi:10.1007/s12028-014-0061-1
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109. Rangel-Castilla L, Gasco J, Nauta HJW, Okonkwo DO, Robertson CS. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus. 2008;25(4). doi:10.3171/FOC.2008.25.10.E7
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. Donnelly J, Czosnyka M, Adams H, et al. Individualizing Thresholds of Cerebral Perfusion Pressure Using Estimated Limits of Autoregulation. Crit Care Med. 2017;45(9):1464-1471. doi:10.1097/CCM.0000000000002575
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. Robba C, Cardim D, Sekhon M, Budohoski K, Czosnyka M. Transcranial Doppler: a stethoscope for the brain-neurocritical care use. J Neurosci Res. 2018;96(4):720-730. doi:10.1002/jnr.24148
- Owens, A.P., 3rd; Mackman, N. Tissue factor and thrombosis: The clot starts here. Thromb. Haemost. 2010, 104, 432–439. Lang EW, Chesnut RM. A Bedside Method for Investigating the Integrity and Critical Thresholds of Cerebral Pressure Autoregulation in Severe Traumatic Brain Injury Patients. Vol 14.; 2000.
- Kenawy, H.I.; Boral, I.; Bevington, A. Complement-Coagulation Cross-Talk: A Potential Mediator of the Physiological Activation of Complement by Low pH. Front. Immunol. 2015, 6, 215. Annane D, Ouanes-Besbes L, de Backer D, et al. A Global Perspective on Vasoactive Agents in Shock.
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: Modulatory effects of hypothermia. J Neuroinflammation. 2010;7. doi:10.1186/1742-2094-7-74
- Bonaventura, A.; Vecchie, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. Zhu Y, Yin H, Zhang R, Ye X, Wei J. Therapeutic hypothermia versus normothermia in adult patients with traumatic brain injury: a meta-analysis. Springerplus. 2016;5(1). doi:10.1186/s40064-016-2391-2
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. Hirst TC, Klasen MG, Rhodes JK, MacLeod MR, Andrews PJD. A Systematic Review and Meta-Analysis of Hypothermia in Experimental Traumatic Brain Injury: Why Have Promising Animal Studies Not Been Replicated in Pragmatic Clinical Trials? J Neurotrauma. 2020;37(19):2057-2068. doi:10.1089/neu.2019.6923
- Kambas, K.; Markiewski, M.M.; Pneumatikos, I.A.; Rafail, S.S.; Theodorou, V.; Konstantonis, D.; Kourtzelis, I.; Doumas, M.N.; Magotti, P.; Deangelis, R.A.; et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J. Immunol. 2008, 180, 7368–7375. Cooper RJ, Wears RL, Schriger DL. Reporting research results: Recommendations for improving communication. Ann Emerg Med. 2003;41(4):561-564. doi:10.1067/mem.2003.135
- Georg, P.; Astaburuaga-Garcia, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gabel, C.; Schneider, M.; Streitz, M.; et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 2022, 185, 493–512.e425. Polderman KH. Application of therapeutic hypothermia in the intensive care unit: Opportunities and pitfalls of a promising treatment modality - Part 2: Practical aspects and side effects. Intensive Care Med. 2004;30(5):757-769. doi:10.1007/s00134-003-2151-y
- Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Laukova, L.; Weber, V. Activated Platelets and Platelet-Derived Extracellular Vesicles Mediate COVID-19-Associated Immunothrombosis. Front. Cell Dev. Biol. 2022, 10, 914891. Andrews PJD, Sinclair HL, Rodriguez A, et al. Hypothermia for Intracranial Hypertension after Traumatic Brain Injury. New England Journal of Medicine. 2015;373(25):2403-2412. doi:10.1056/NEJMoa1507581
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. Zwerus R, Absalom A. Update on anesthetic neuroprotection. Curr Opin Anaesthesiol. 2015;28(4):424-430. doi:10.1097/ACO.0000000000000212
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. Almaas R, Saugstad OD, Pleasure D, Rootwelt T. Effect of Barbiturates on Hydroxyl Radicals, Lipid Peroxidation, and Hypoxic Cell Death in Human NT2-N Neurons. Vol 92.; 2000.
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. Léger M, Frasca D, Roquilly A, et al. Early use of barbiturates is associated with increased mortality in traumatic brain injury patients from a propensity score-based analysis of a prospective cohort. PLoS One. 2022;17(5):e0268013. doi:10.1371/journal.pone.0268013
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. Majdan M, Mauritz W, Wilbacher I, Brazinova A, Rusnak M, Leitgeb J. Barbiturates use and its effects in patients with severe traumatic brain injury in five European countries. J Neurotrauma. 2013;30(1):23-29. doi:10.1089/neu.2012.2554
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. Roberts I, Sydenham E. Barbiturates for acute traumatic brain injury. Cochrane Database of Systematic Reviews. Published online December 12, 2012. doi:10.1002/14651858.cd000033.pub2
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. Stover JF, Lenzlinger PM, Stocker R, et al. Thiopental in CSF and serum correlates with prolonged loss of cortical activity. Eur Neurol. 1998;39(4):223-228. doi:10.1159/000007938
- Initiative, C.-H.G. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. Wheeler DW, Thompson AJ, Corletto F, et al. Anaesthetic impairment of immune function is mediated via GABAA receptors. PLoS One. 2011;6(2). doi:10.1371/journal.pone.0017152
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. Loop T, Humar M, Pischke S, et al. Thiopental Inhibits Tumor Necrosis Factor-Induced Activation of Nuclear Factor B through Suppression of IB Kinase Activity. Vol 99.; 2003. http://pubs.asahq.org/anesthesiology/article-pdf/99/2/360/408372/0000542-200308000-00017.pdf
- Koning, R.; Bastard, P.; Casanova, J.L.; Brouwer, M.C.; van de Beek, D.; Amsterdam, U.M.C. COVID-19 Biobank Investigators. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. 2021, 47, 704–706. Andrefsky, J.C.; Frank, J.I.; Chyatte, D.; The ciliospinal reflex in pentobarbital coma. J Neurosurg. 1999;90:644-646.
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. Cairns CJ, Thomas B, Fletcher S, Parr MJ, Finfer SR. Life-threatening hyperkalaemia following therapeutic barbiturate coma. Intensive Care Med. 2002;28(9):1357-1360. doi:10.1007/s00134-002-1399-y
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020, 383, 2451–2460. Aytuluk HG, Topcu H. Severe hypokalemia and rebound hyperkalemia during barbiturate coma in patients with severe traumatic brain injury. Neurocirugia. 2020;31(5):216-222. doi:10.1016/j.neucir.2019.12.003
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA 2020, 234, 1565–1567. Ellington AL. Electroencephalography and Clinical Neurophysiology CLINICAL AND LABORATORY NOTES ELECTROENCEPHALOGRAPHIC PATTERN OF BURST SUPPRESSION IN A CASE OF BARBITURATE COMA.
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. Hutchinson PJ, Kolias AG, Timofeev IS, et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. New England Journal of Medicine. 2016;375(12):1119-1130. doi:10.1056/nejmoa1605215
- Wilson JG; COVID-19-Related ARDS: Key Mechanistic Features and Treatments. Crit Care 2020, 24, 102, 10.3390/jcm11164896.Cooper DJ, Rosenfeld J v., Murray L, et al. Decompressive Craniectomy in Diffuse Traumatic Brain Injury. New England Journal of Medicine. 2011;364(16):1493-1502. doi:10.1056/nejmoa1102077.
- doi:10.1164/ajrccm.158.1.9708031; Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? . Am J Respir Crit Care Med 1998, 158, 3, doi:10.1164/ajrccm.158.1.9708031.
- Calfee CS; Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials.. Lancet Respir Med 2014, 2, 611, doi:10.1016/S2213-2600(14)70097-9.
- Satturwar S; Postmortem Findings Associated With SARS-CoV-2: Systematic Review and Meta-analysis.. Am J Surg Pathol 2021, 45, 587, doi:10.1097/PAS.0000000000001650.
- Katzenstein AL; Diffuse alveolar damage--the role of oxygen, shock, and related factors. A review. . Am J Pathol 1976, 85, 209.
- Tomashefski JF; The pulmonary vascular lesions of the adult respiratory distress syndrome.. Am J Pathol 1983, 112, 112.
- Hariri LP; Lung Histopathology in Coronavirus Disease 2019 as Compared With Severe Acute Respiratory Sydrome and H1N1 Influenza: A Systematic Review. . Chest 2021, 159, 73, 10.1016/j.chest.2020.09.259.
- Milross L; Post-mortem lung tissue: the fossil record of the pathophysiology and immunopathology of severe COVID-19.. Lancet Respir Med 2022, 10, 95, doi:10.1016/S2213-2600(21)00408-2.
- Carsana L.; Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. . Lancet Infect Dis 2020, 20, 1135, 10.1016/S1473-3099(20)30434-5.
- Satturwar S; Postmortem Findings Associated With SARS-CoV-2: Systematic Review and Meta-analysis.. Am J Surg Pathol 2021, 45, 87, doi:10.1097/PAS.0000000000001650.
- Ackermann M; Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med 2020, 383, 120, 10.1056/NEJMoa2015432.
- Poissy J; Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184, 10.1161/CIRCULATIONAHA.120.047430.
- Helms J; High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 2020, 46, 1089, 10.1007/s00134-020-06062-x.
- Villalba JA; Vasculopathy and Increased Vascular Congestion in Fatal COVID-19 and ARDS. Am J Respir Crit Care Med 2022, 10.1164/rccm.202109-2150OC, in press, 10.1164/rccm.202109-2150OC.
- Santamarina MG; COVID-19: What Iodine Maps From Perfusion CT can reveal-A Prospective Cohort Study. Crit Care 2020, 24, 19, doi:10.1186/s13054-020-03333-3.
- Li Q; CT features of coronavirus disease 2019 (COVID-19) with an emphasis on the vascular enlargement pattern. Eur J Radiol 2021, 134, 109442, 10.1016/j.ejrad.2020.109442.
- Poschenrieder F; Severe COVID-19 pneumonia: Perfusion analysis in correlation with pulmonary embolism and vessel enlargement using dual-energy CT data. PLoS One 2021, 16, e0252478, doi:10.1371/journal.pone.0252478.
- Patel BV; Pulmonary Angiopathy in Severe COVID-19: Physiologic, Imaging, and Hematologic Observations. Am J Respir Crit Care Med 2020, 202, 690, 10.1164/rccm.202004-1412OC.
- Gattinoni L; Anatomical and functional intrapulmonary shunt in acute respiratory distress syndrome. Crit Care Med 2008, 36, 669, 10.1097/01.CCM.0000300276.12074.E1.
- Gattinoni L; Anatomical and functional intrapulmonary shunt in acute respiratory distress syndrome. Crit Care Med 2008, 36, 669, 10.1097/01.CCM.0000300276.12074.E1.
- Chiumello D; Physiological and quantitative CT-scan characterization of COVID-19 and typical ARDS: a matched cohort study. Intensive Care Med 2020, 46, 2187, 10.1007/s00134-020-06281-2.
- Barbeta E; SARS-CoV-2-induced Acute Respiratory Distress Syndrome: Pulmonary Mechanics and Gas-Exchange Abnormalities. Ann Am Thorac Soc 2020, 17, 1164, 10.1513/AnnalsATS.202005-462RL.
- Vasques F; Physiological dead space ventilation, disease severity and outcome in ventilated patients with hypoxaemic respiratory failure due to coronavirus disease 2019. Intensive Care Med 2020, 46, 2092, 10.1007/s00134-020-06197-x..
- Camporota L; Prone Position in COVID-19 and -COVID-19 Acute Respiratory Distress Syndrome: An International Multicenter Observational Comparative Study. Crit Care Med 2022, 50, 633, 10.1097/CCM.0000000000005354.
- Grieco DL; Respiratory physiology of COVID-19-induced respiratory failure compared to ARDS of other etiologies. Crit Care 2020, 24, 529, 10.1186/s13054-020-03253-2.
- Kummer RL; Paradoxically Improved Respiratory Compliance With Abdominal Compression in COVID-19 ARDS. Chest 2021, 160, 1739, 10.1016/j.chest.2021.05.012.
- Haudebourg AF; Respiratory Mechanics of COVID-19- versus Non-COVID-19-associated Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2020, 202, 287, 10.1164/rccm.202004-1226LE.
- Panwar R; Compliance Phenotypes in Early Acute Respiratory Distress Syndrome before the COVID-19 Pandemic. Am J Respir Crit Care Med 2020, 202, 1244, 10.1164/rccm.202005-2046OC.
- Beloncle F; Longitudinal changes in compliance, oxygenation and ventilatory ratio in COVID-19 versus non-COVID-19 pulmonary acute respiratory distress syndrome. Crit Care 2021, 25, 248, 10.1186/s13054-021-03665-8.
- Gattinoni L.; Reply by Gattinoni et al. to Hedenstierna et al., to Maley et al., to Fowler et al., to Bhatia and Mohammed, to Bos, to Koumbourlis and Motoyama, and to Haouzi et al. Am J Respir Crit Care Med 2020, 202, 628, 10.1164/rccm.202004-1052LE.
- Marini JJ; Energetics and the Root Mechanical Cause for Ventilator-induced Lung Injury. Anesthesiology 2018, 128, 1062, doi:10.1097/ALN.0000000000002203.
- Marini JJ; Static and Dynamic Contributors to Ventilator-induced Lung Injury in Clinical Practice. Pressure, Energy, and Power. Am J Respir Crit Care Med 2020, 201, 767, 10.1164/rccm.201908-1545CI.
- Fan E; An Official American Thoracic Society/European Society of Intensive Care Medicine/Society of Critical Care Medicine Clinical Practice Guideline: Mechanical Ventilation in Adult Patients with Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2017, 195, 1253, 10.1164/rccm.201703-0548ST.
- Alhazzani W; Surviving Sepsis Campaign: Guidelines on the Management of Critically Ill Adults with Coronavirus Disease 2019 (COVID-19). Crit Care Med 2008, 28, e440, 10.1097/CCM.0000000000004363.
- Dreyfuss D; Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998, 157, 294, 10.1164/ajrccm.157.1.9604014.
- Stewart TE; Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N Engl J Med 1998, 338, 355, 10.1056/NEJM199802053380603.
- Brochard L; Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. The Multicenter Trail Group on Tidal Volume reduction in ARDS. Am J Respir Crit Care Med 1998, 158, 1831, 10.1164/ajrccm.158.6.9801044.
- Brower RG; Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit Care Med 1999, 27, 1492, 10.1097/00003246-199908000-00015.
- Liu X; Ventilatory Ratio in Hypercapnic Mechanically Ventilated Patients with COVID-19-associated Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2020, 201, 1297, 10.1164/rccm.202002-0373LE.
- Marini JJ; Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329, 10.1001/jama.2020.6825.
- Gattinoni L; Intensive Care Med . Intensive Care Med 2022, 48, 728, 10.1007/s00134-022-06698-x.
- Barthelemy R; Haemodynamic impact of positive end-expiratory pressure in SARS-CoV-2 acute respiratory distress syndrome: oxygenation versus oxygen delivery. Br J Anaesth 2021, 126, e70, 10.1016/j.bja.2020.10.026.
- Suter PM; Optimum end-expiratory airway pressure in patients with acute pulmonary failure. N Engl J Med 1975, 292, 284, 10.1056/NEJM197502062920604.
- Dickel S; Nationwide Cross-Sectional Online Survey on the Treatment of COVID-19-ARDS: High Variance in Standard of Care in German ICUs. J Clin Med 2021, 10, 3363, 10.3390/jcm10153363.
- Grasselli G; Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574, 10.1001/jama.2020.5394.
- Chiumello D; Positive end-expiratory pressure in COVID-19 acute respiratory distress syndrome: the heterogeneous effects. Crit Care 2021, 25, 431, 10.1186/s13054-021-03839-4.
- Sinha P; Analysis and Clinical Performance of the Ventilatory Ratio in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2019, 199, 333, 10.1164/rccm.201804-0692OC.
- Beitler JR; Effect of Titrating Positive End-Expiratory Pressure (PEEP) With an Esophageal Pressure-Guided Strategy vs an Empirical High PEEP-Fio2 Strategy on Death and Days Free From Mechanical Ventilation Among Patients With Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA 2019, 321, 846, 10.1001/jama.2019.0555.
- Pan C; Lung Recruitability in COVID-19-associated Acute Respiratory Distress Syndrome: A Single-Center Observational Study. Am J Respir Crit Care Med 2020, 201, 1294, 10.1164/rccm.202003-0527LE.
- Ball L; Computed tomography assessment of PEEP-induced alveolar recruitment in patients with severe COVID-19 pneumonia. Crit Care 2021, 25, 81, 10.1186/s13054-021-03477-w.
- Protti A; Lung Response to a Higher Positive End-Expiratory Pressure in Mechanically Ventilated Patients With COVID-19. Chest 2022, 161, 979, 10.1016/j.chest.2021.10.012.
- Roesthuis L; Advanced respiratory monitoring in COVID-19 patients: use less PEEP! . Crit Care 2020, 24, 230, 1186/s13054-020-02953-z.
- Perier F; Effect of Positive End-Expiratory Pressure and Proning on Ventilation and Perfusion in COVID-19 Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2020, 202, 1713, doi:10.1164/rccm.202008-3058LE.
- Mauri T; Potential for Lung Recruitment and Ventilation-Perfusion Mismatch in Patients With the Acute Respiratory Distress Syndrome From Coronavirus Disease 2019. Crit Care Med 2020, 48, 1129, 10.1097/CCM.0000000000004386.
- Chen L; Potential for Lung Recruitment Estimated by the Recruitment-to-Inflation Ratio in Acute Respiratory Distress Syndrome. A Clinical Trial. Am J Respir Crit Care Med 2020, 201, 178, 10.1164/rccm.201902-0334OC.
- Gattinoni L; Prone Position and COVID-19: Mechanisms and Effects. . Crit Care Med 2022, 50, 873, 10.1097/CCM.0000000000005486.
- Rouby J.J; Acute respiratory distress syndrome: lessons from computed tomography of the whole lung. Crit Care Med 2003, 31, 5285, 10.1097/01.CCM.0000057905.74813.BC.
- Albert RK; The prone position eliminates compression of the lungs by the heart. Am J Respir Crit Care Med 2000, 161, 1660, 10.1164/ajrccm.161.5.9901037.
- Pelosi P; Vertical gradient of regional lung inflation in adult respiratory distress syndrome. Am J Respir Crit Care Med 1994, 149, 8, 10.1164/ajrccm.149.1.8111603.
- Gattinoni L; Prone position in acute respiratory distress syndrome. Rationale, indications, and limits. Am J Respir Crit Care Med 2013, 188, 1286, 10.1164/rccm.201308-1532CI.
- Guerin C; Prone position in ARDS patients: why, when, how and for whom. Intensive Care Med 2020, 46, 2385, 10.1007/s00134-020-06306-w.
- Mentzelopoulos SD; Prone position reduces lung stress and strain in severe acute respiratory distress syndrome. Eur Respir J 2005, 25, 534, 10.1183/09031936.05.00105804.
- Langer T; Prone position in intubated, mechanically ventilated patients with COVID-19: a multi-centric study of more than 1000 patients. Crit Care 2021, 25, 128, 10.1186/s13054-021-03552-2.
- Bellani G; The LUNG SAFE study: a presentation of the prevalence of ARDS according to the Berlin Definition! . Crit Care 2016, 20, 268, 10.1186/s13054-016-1443-x.
- Guerin C; Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013, 368, 2159, 10.1056/NEJMoa1214103.
- Mathews KS; Prone Positioning and Survival in Mechanically Ventilated Patients With Coronavirus Disease 2019-Related Respiratory Failure. Crit Care Med 2021, 49, 1026, 10.1097/CCM.0000000000004938.
- Fossali T; Effects of Prone Position on Lung Recruitment and Ventilation-Perfusion Matching in Patients With COVID-19 Acute Respiratory Distress Syndrome: A Combined CT Scan/Electrical Impedance Tomography Study. Crit Care Med 2022, 50, 723, 10.1097/CCM.0000000000005450.
- Zarantonello F; Early physiological effects of prone positioning in COVID-19 Acute Respiratory Distress Syndrome. Anesthesiology 2022, 137, 327, 10.1097/ALN.0000000000004296.
- Rossi S; Mechanisms of oxygenation responses to proning and recruitment in COVID-19 pneumonia. Intensive Care Med 2022, 48, 56, 10.1007/s00134-021-06562-4.
- Protti A; Lung response to prone positioning in mechanically-ventilated patients with COVID-19.. Crit Care 2022, 26, 127, 10.1186/s13054-022-03996-0..
- Iwashyna TJ; Implications of Heterogeneity of Treatment Effect for Reporting and Analysis of Randomized Trials in Critical Care. Am J Respir Crit Care Med 2015, 192, 1045, 10.1164/rccm.201411-2125CP.