Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 1 by Mohit Bansal.
Neuroblastoma is a pediatric cancer responsible for approximately 15% of all childhood cancer deaths. Aberrant MYCN activation, as a result of genomic MYCN amplification, is a major driver of high-risk neuroblastoma, which has an overall survival rate of less than 50%, despite the best treatments currently available. Metabolic reprogramming is an integral part of the growth-promoting program driven by MYCN, which fuels cell growth and proliferation by increasing the uptake and catabolism of nutrients, biosynthesis of macromolecules, and production of energy. This reprogramming process also generates metabolic vulnerabilities that can be exploited for therapy.
- cancer metabolism
- metabolic reprogramming
- MYCN
1. Aerobic Glycolysis in Neuroblastoma
Aerobic glycolysis, also known as the Warburg effect, describes the metabolic phenotype of increased glucose uptake and lactate production in the presence of abundant oxygen [58,59,60,61][1][2][3][4] (Figure 1). It has long been recognized that neuroblastoma tumors display increased glucose uptake, as determined by 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) imaging [62][5]. In addition, it has been shown that increased glycolysis promotes the survival and growth of neuroblastoma cells in vitro and in vivo [63][6]. Both MYCN and hypoxia-inducible factor 1α (HIF-1α) are required for sustaining the aerobic glycolysis phenotype in neuroblastoma cells by transcriptional upregulation of glycolytic enzymes, including hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA) [64][7]. More recent studies further confirm a critical role for MYCN in promoting glycolysis in neuroblastoma cells [65,66][8][9]. These findings are consistent with the extensive evidence for MYC as a key transcriptional activator of genes involved in glycolysis [67][10].
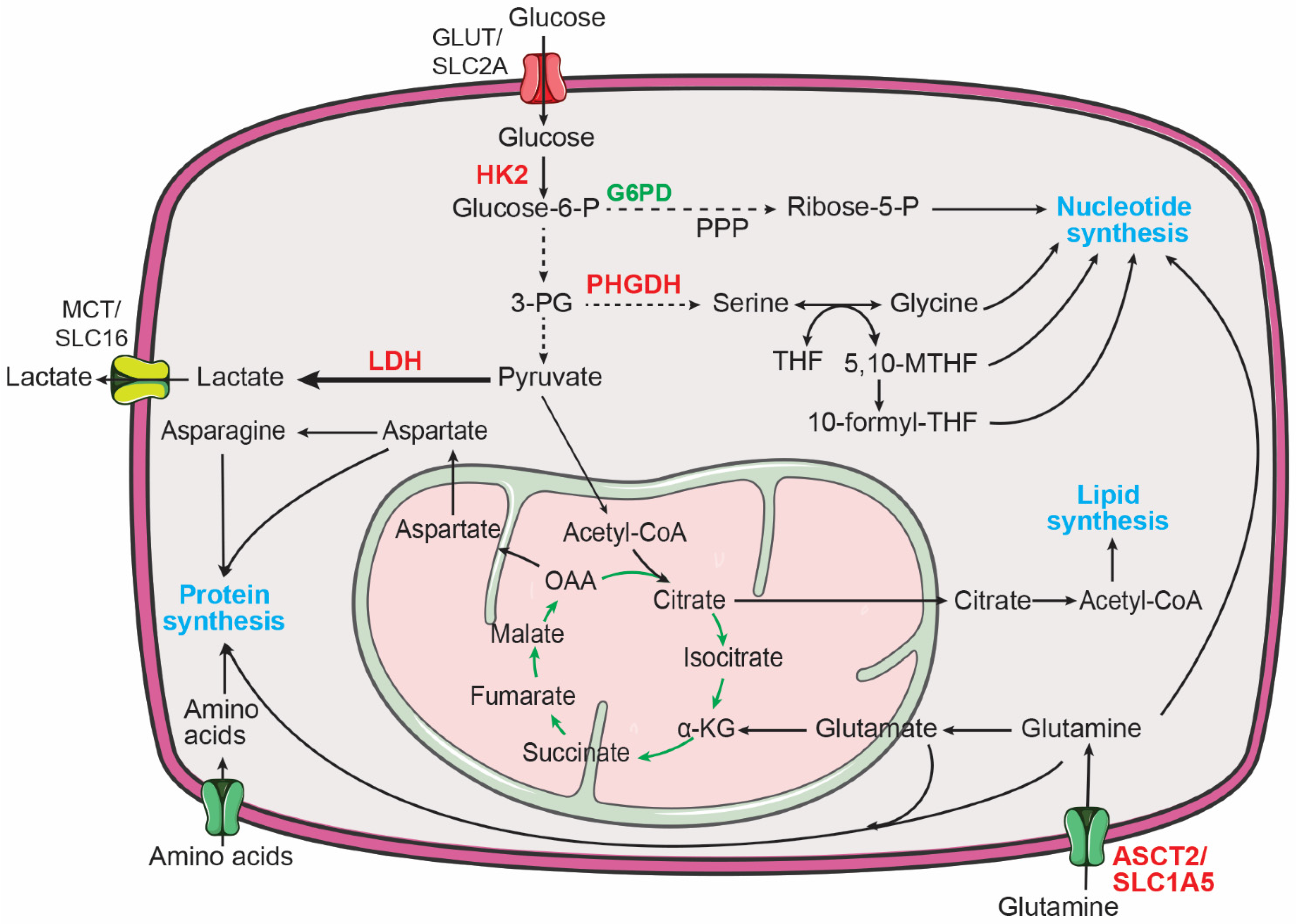
Figure 1. Major metabolic pathways for supporting macromolecule biosynthesis in cancer cells. Increased central carbon metabolism provides building blocks for the production of proteins, nucleotides, and lipids to sustain the proliferation of cancer cells. 3-PG, 3-phosphoglycerate; 5,10-MTHF, 5,10-methylene-tetrahydrofolate; 10-formyl-THF, 10-formyl-tetrahydrofolate; α-KG, α-ketoglutarate; Glucose-6-P, glucose-6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; HK2, hexokinase 2; LDH, lactate dehydrogenase; OAA, oxaloacetate; PHGDH, phosphoglycerate dehydrogenase; PPP, pentose phosphate pathway; THF, tetrahydrofolate. MYCN targets are highlighted in red.
Interestingly, recent studies from the Tong laboratory revealed novel mechanisms underlying the increased glycolysis in neuroblastoma cells [68,69][11][12]. Through a combination of bioinformatics and overexpression and knockdown studies, Li et al. identified CUT-like homeobox 1 (CUX1) as a transcription factor that upregulates glycolytic genes to promote aerobic glycolysis in neuroblastoma cells. Moreover, they showed that circ-CUX1, a circular RNA generated from CUX1 exon 2 and part of intron 2, can interact with EWS RNA-binding protein 1 (EWSR1) to facilitate EWSR1 interaction with MYC-associated zinc finger protein (MAZ). The EWSR1-MAZ complex, in turn, transcriptionally upregulates CUX1 expression. Disrupting this circ-CUX1/EWSR1/MAZ axis suppresses glycolysis and the growth and metastasis of neuroblastoma cells in vivo [68][11]. It is noteworthy to mention that high circ-CUX1 and CUX1 expression in neuroblastoma tumors and cell lines is not associated with MYCN amplification [68][11], suggesting that this is an MYCN-independent mechanism for promoting glycolysis in neuroblastoma cells. In another study, it was reported that hepatocyte nuclear factor 4 alpha (HNF4A) and its derived long-noncoding RNA (HNF4A-AS1) promote aerobic glycolysis in neuroblastoma cells by upregulating HK2 and the major glucose transporter SLC2A1 (also known as GLUT1) [69][12]. Moreover, it was found that high HNF4A-AS1 expression is associated with MYCN amplification in neuroblastoma tumors and that MYCN overexpression or knockdown in neuroblastoma cell lines increases or downregulates HNF4A-AS1 expression, respectively [69][12]. These findings reveal an MYCN-dependent mechanism for promoting glucose import to sustain glycolysis in neuroblastoma cells.
The conversion of pyruvate to lactate is catalyzed by lactate dehydrogenase (LDH) (Figure 1). Human cells express three LDH isoforms, LDHA, LDHB, and LDHC [70,71][13][14]. A study by Qing et al. showed that LDHA expression is increased in MYCN-amplified neuroblastoma tumors and that MYCN is required for maintaining LDHA expression in neuroblastoma cells [64][7]. However, a later study by Dorneburg et al. provided evidence that LDHA expression is independent of the MYCN amplification status and MYCN expression levels, as determined by a combination of gene expression profiling, immunohistochemistry of neuroblastoma tumors, and MYCN overexpression studies [72][15]. Nevertheless, both studies showed that LDHA is required for neuroblastoma cell growth and tumorigenicity. Interestingly, the study by Dorneburg et al. reported that knockout of LDHA has no significant effect on glucose consumption and lactate production in neuroblastoma cells in culture and in xenograft tumors. Furthermore, knockdown of LDHB expression in LDHA knockout cells also fails to produce a significant effect on glucose consumption and lactate production. Of note, these neuroblastoma cells do not express significant levels of LDHC. These findings suggest a functional role for LDHA and LDHB in neuroblastoma, independent of aerobic glycolysis [72][15], but also raise the provocative question of how lactate production is maintained in neuroblastoma cells with no significant levels of LDH expression. Stable isotope tracing experiments using uniformly labeled 13C-glucose may help address the source of lactate in these LDH-depleted neuroblastoma cells and underlying mechanisms.
2. Reprogramming of Amino Acid Metabolism in Neuroblastoma
Of the 20 proteinogenic amino acids, 9 cannot be synthesized by humans and, thus, are considered essential amino acids, including histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. These essential amino acids can enter cells via amino acid transport [73][16]. It has been reported that MYCN can transcriptionally activate the expression of the solute carrier family (SLC) of genes, SLC7A5 and SLC43A1, through direct binding to the E-box sequence within both genes. Knockdown of SLC7A5 or SLC43A1 expression in neuroblastoma cells represses the uptake of essential amino acids, as determined by 3H-Leucine transport assay, leading to a significant decrease in the intracellular levels of isoleucine, leucine, phenylalanine, and valine. Moreover, SLC7A5 or SLC43A1 depletion reduces MYCN expression by interfering with MYCN mRNA translation and attenuates neuroblastoma cell growth in vitro and in vivo. These findings suggest that MYCN and SLC7A5/SLC43A1 form a positive feedback loop to amplify their expression for sustaining the growth and tumorigenicity of neuroblastoma cells [74][17].
However, amino acid metabolic reprogramming in cancer cells is primarily concerned with the eleven non-essential amino acids, including alanine, arginine, asparagine, aspartate, cysteine, glutamate, glutamine, glycine, proline, serine, and tyrosine [75][18]. Activating transcription factor 4 (ATF4) is a master transcriptional regulator of amino acid synthesis and transport [76,77,78][19][20][21]. In neuroblastoma cells, ATF4 cooperates with the histone lysine demethylase KDM4C to transcriptionally upregulate the expression of enzymes for synthesis of eight non-essential amino acids, including alanine, arginine, asparagine, aspartate, cysteine, glutamate, glycine, and serine. ATF4 and KDM4C also cooperate to induce mRNA expression of SLC1A4, SLC1A5, SLC3A2, SLC6A9, SLC7A1, SLC7A5, and SLC7A11, which are involved in the transport of most amino acids [79][22]. Analysis of patient data reveals significantly higher ATF4 mRNA levels in neuroblastoma tumors with MYCN amplification [80][23] and higher ATF4 expression is associated with poor prognosis in neuroblastoma patients [81][24]. Further investigation demonstrates that MYCN binds to the promoter of ATF4 to increase its mRNA expression [80][23]. Together, these findings suggest that the MYCN-ATF4 axis, in cooperation with epigenetic regulators, including KDM4C, is a major mechanism to increase the pools of amino acids for sustaining the MYCN-mediated growth program in neuroblastoma [80,82][23][25].
2.1. Glutamine Metabolism
It has long been established that glutamine is critical for the growth of cancer cells [83,84,85,86][26][27][28][29]. Highly proliferating cancer cells show predominantly aerobic glycolysis, in which most of the glycolytic pyruvate is converted to lactate instead of feeding into the TCA cycle [58,59,60][1][2][3]. As a result, cancer cells must find other sources of anaplerosis to sustain the TCA cycle for producing ATP and for supplying intermediates for macromolecule biosynthesis. Glutamine, as the most abundant circulating amino acid in the blood and tissues [87][30], is a major source of anaplerosis [88][31]. It has a five-carbon backbone and two nitrogen atoms, which serves as a carbon donor to drive the TCA cycle via anaplerosis and a nitrogen donor for biosynthesis of nucleotides and non-essential amino acids [89,90,91][32][33][34] (Figure 2). Increased glutamine metabolism has been considered a “hallmark of cancer metabolism” [89][32].
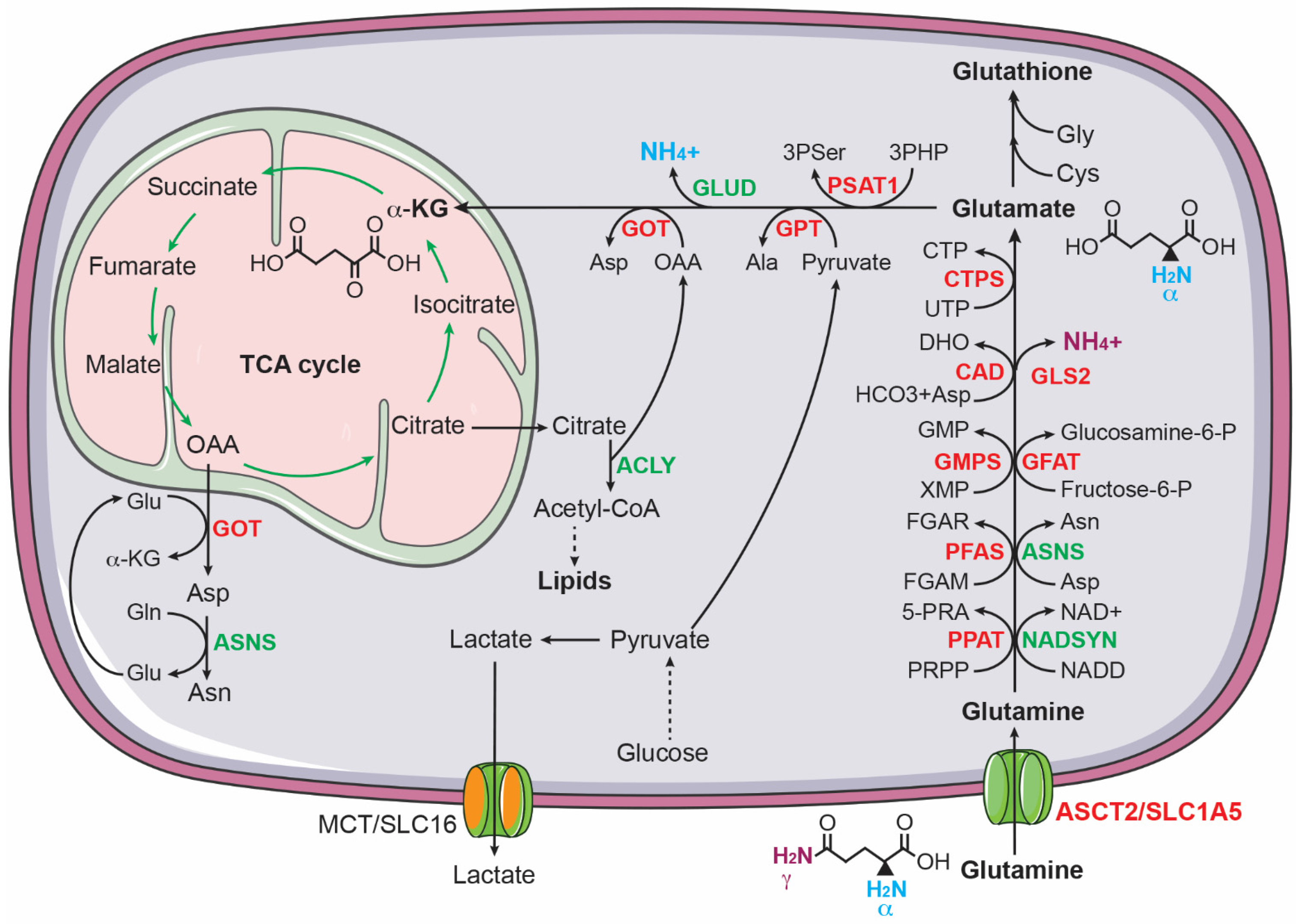
Figure 2. Glutamine uptake and catabolism supports biosynthesis in cancer cells. Glutamine donates its γ-nitrogen for the biosynthesis of asparagine (Asn), nucleotides, NAD+, and glucosamine-6-phosphate (glucosamine-6-P) and its α-nitrogen for 3-phospho-serine (3Pser), alanine (Ala), and aspartate (Asp). Glutamine also provides carbon via α-ketoglutarate to sustain the TCA cycle for producing ATP and intermediates for the biosynthesis of aspartate, asparagine, and lipids. ASNS, asparagine synthetase; CAD, carbamoyl phosphate synthase, aspartate transcarbamoylase, and dihydroorotase; CTPS, CTP synthase; GFAT, glutamine fructose-6-phosphate amidotransferase; GMPS, guanine monophosphate synthase; GOT, glutamic-oxaloacetic transaminase; GPT, glutamic--pyruvic transaminase; NADSYN, NAD synthetase; PFAS, phosphoribosyl formylglycinamidine synthase; PPAT, phosphoribosyl pyrophosphate amidotransferase. MYCN targets are highlighted in red.
Enhanced glutamine metabolism is crucial for the survival and proliferation of neuroblastoma cells. Qing et al. reported that glutamine deprivation triggers apoptosis in MYCN-amplified neuroblastoma cell lines by inducing pro-apoptotic proteins PUMA, NOXA, and TRIB3 [92][35]. Subsequent investigation revealed that MYCN-amplified neuroblastoma cells express high levels of ASCT2 (also known as SLC1A5), which functions as a major transporter for uptake of glutamine in MYCN-amplified neuroblastoma cells. Increased ASCT2 expression is associated with advanced stages of neuroblastoma and poor prognosis in neuroblastoma patients. The upregulation of ASCT2 is mediated by MYCN and ATF4, which bind the ASCT2 promoter to induce its transcription. Knockdown of ASCT2 expression markedly inhibited glutamine uptake and decreased the intracellular levels of downstream metabolites of glutaminolysis, including glutamate and the TCA cycle intermediates succinate and fumarate. Moreover, ASCT2 knockdown induced G1 arrest and apoptosis in MYCN-amplified neuroblastoma cell lines and significantly inhibited their ability to form a tumor in immunodeficient mice [93][36].
In addition to promoting glutamine uptake, MYCN activates glutaminolysis in neuroblastoma cells [94][37], which is a process of replenishing the TCA cycle by converting glutamine to α-ketoglutarate [89,90,91][32][33][34]. During glutaminolysis, glutamine is first converted to glutamate by removing its γ-amino group, which is commonly catalyzed by glutaminases (GLS in the cytosol and GLS2 in the mitochondria). It has been shown that MYCN stimulates the conversion of glutamine to glutamate in MYCN-amplified neuroblastoma cells by directly activating GLS2 transcription [95][38]. Alternatively, glutamine can be converted to glutamate through amino acid and nucleotide synthesis pathways. Asparagine synthetase (ASNS), a direct transcriptional target of ATF4 [76][19], converts glutamine and aspartate to glutamate and asparagine. In purine synthesis, two glutamine molecules donate two nitrogen atoms for the synthesis of one molecule of inosine monophosphate (IMP), generating two molecules of glutamate. The synthesis of guanosine monophosphate (GMP) from IMP requires an additional nitrogen atom from glutamine, producing one more molecule of glutamate. Pyrimidine synthesis generates two glutamate molecules: the first step consumes one nitrogen atom from glutamine and the synthesis of CTP from UTP needs one more nitrogen atom from glutamine. All these reactions in purine and pyrimidine nucleotide synthesis are catalyzed by glutamine amidotransferases (PPAT, PFAS, GMPS, CAD, and CTPS1/2), which are transcriptionally activated by MYCN in neuroblastoma cells [94,96][37][39]. The functional significance of these enzymatic reactions is underscored by the observation that overexpression of MYCN markedly sensitizes non-MYCN-amplified neuroblastoma cells to the glutamine analogue DON (6-diazo-5-oxo-L-norleucine), which blocks the conversion of glutamine to glutamate by inhibiting glutamine amidotransferases [66][9].
Following the conversion of glutamine to glutamate, the next step in utilizing glutamine carbon for cellular bioenergetic and biosynthetic needs is to convert glutamate to α-ketoglutarate to replenish the TCA cycle. Through the TCA cycle, glutamine-derived carbon can be used to drive NADH and FDAH2 production and to generate citrate and oxaloacetate. Citrate is a source of acetyl-CoA for the biosynthesis of fatty acids and cholesterol and oxaloacetate is a substrate for the biosynthesis of aspartate [89,90,91][32][33][34]. The conversion of glutamate to α-ketoglutarate is catalyzed by glutamate dehydrogenases (GLUD1 and GLUD2) or transaminases, including glutamate-oxaloacetate transaminase (GOT), glutamate-pyruvate transaminase (GPT), and phosphoserine transaminase 1 (PSAT1). These transaminases use the nitrogen atom from glutamate for the biosynthesis of the non-essential amino acids aspartate (GOT and GOT2), alanine (GPT and GPT2), and serine and glycine (PSAT1). MYCN, ATF4, and KDM4C all contribute to transcriptional upregulation of GOT1, GOT2, GPT, GPT2, and PSAT1 in neuroblastoma cells [80,82,94][23][25][37].
In addition to enhancing glutamine transport and glutaminolysis, MYCN may promote glutamine synthesis in neuroblastoma cells. Using MYCN-inducible expression neuroblastoma cell lines and stable isotope tracing with U-13C5-glucose, Oliynyk et al. showed that MYCN induction increases glutamine synthesis from glucose via α-ketoglutarate [65][8]. The functional significance of the MYCN-driven glutamine synthesis in neuroblastoma cells remains to be determined.
Together, these findings suggest that MYCN, ATF4, and KDM4C work together to promote glutamine uptake and metabolism in MYCN-amplified neuroblastoma cells to meet the biosynthetic demands of cell growth and proliferation.
2.2. Serine and Glycine Metabolism
It was reported more than 60 years ago that serine is important in supporting the growth of human cancer cells in a minimal medium supplemented with dialyzed serum [97][40]. Cells can obtain serine via transport and de novo biosynthesis. Upregulation of serine and glycine synthesis is an integral part of cancer metabolism [98,99,100,101][41][42][43][44]. The serine-glycine synthesis pathway consists of four reactions that are catalyzed sequentially by PHGDH, PSAT1, phosphoserine phosphatase (PSPH), and serine hydroxymethyltransferase (SHMT1 in the cytosol and SHMT2 in the mitochondrion). This metabolic pathway links the glycolytic intermediate 3-phosphoglycerate (3PG) to the production of serine, glycine, and the one-carbon carrier 5,10-methylenetetrahydrofolate (5,10-MTHF), along with NADH and α-ketoglutarate (Figure 3). These products are involved in many cellular processes, essential for the growth of cancer cells, including macromolecule synthesis (proteins, nucleic acids, and lipids), redox homeostasis, and methylation regulation. Serine is required for the synthesis of cysteine and sphingolipids; glycine contributes carbon and nitrogen atoms to purine synthesis and is a component of glutathione; and 5,10-MTHF donates carbon to the synthesis of thymidylate and S-adenosylmethionine, the universal methyl donor for methylation reactions. In addition, 5,10-MTHF is a precursor for the synthesis of 10-formyl-THF that donates carbon to purine synthesis [98,99,100,101][41][42][43][44].
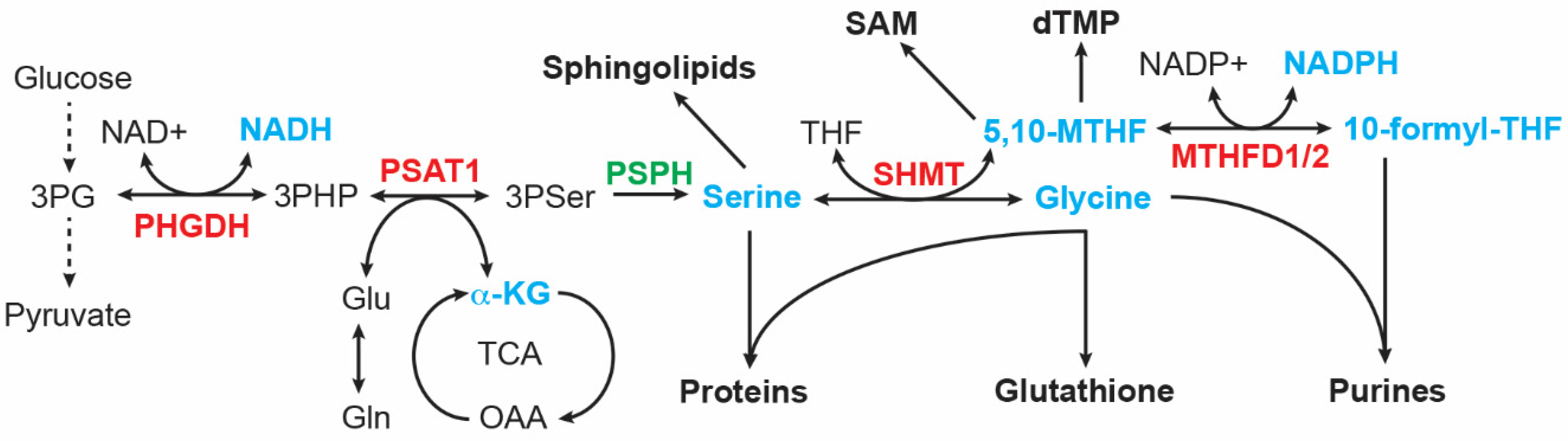
Figure 3. Serine-glycine synthesis pathway. Intermediates and products (highlighted in cyan) of this metabolic pathway serve as building blocks for the synthesis of proteins, lipids, nucleotides, reducing equivalents (NADH and NADPH), glutathione, and S-adenosyl-methionine (SAM). MTHFD, Methylenetetrahydrofolate dehydrogenase; PHGDH, phosphoglycerate dehydrogenase; PSAT1, phosphoserine Aminotransferase 1; PSPH, phosphoserine phosphatase; SHMT, serine hydroxymethyltransferase. MYCN targets are highlighted in red.
Neuroblastoma tumors with MYCN amplification show increased expression of serine and glycine synthesis enzymes, including PHGDH, PSAT1, and SHMT2. The increased expression is primarily the result of transcriptional activation, mediated by both MYCN and ATF4. These two transcription factors can directly bind the promoters of serine-glycine pathway genes to activate their transcription. Moreover, MYCN and ATF4 form a positive feedback loop, with MYCN activating ATF4 mRNA expression and ATF4 stabilizing MYCN protein by suppressing FBXW7-mediated MYCN ubiquitination and degradation [80][23]. This positive feedback loop presumably reinforces the hyperactive transcription state of these genes to boost production outputs of the serine-glycine synthesis pathway.
ATF4 also cooperates with enzymes that control histone methylation states for transcriptional activation of serine-glycine pathway genes in neuroblastoma cells. G9A (also known as KMT1C) is an H3K9 methyltransferase that has a major role in catalyzing histone H3 lysine 9 monomethylation (H3K9me1) and H3K9me2 in euchromatin [102,103,104,105][45][46][47][48]. H3K9me1 is an active mark and H3K9me2 a repressive mark [106,107][49][50]. It has been reported that G9A is essential for maintaining serine-glycine pathway genes in an active state marked by H3K9me1 [81][24]. G9A knockdown or inhibition represses the expression of serine-glycine synthesis enzymes, leading to a significant decrease in intracellular serine and glycine levels and autophagic cell death in neuroblastoma cells. This cell death phenotype can be rescued by supplemental serine. Higher G9A expression, which is associated with poor prognosis in neuroblastoma patients, increases serine production from glucose and enhances the proliferation and tumorigenicity of neuroblastoma cells. Importantly, ATF4 is required for G9A to upregulate the serine-glycine pathway. The molecular basis for the ATF4-G9A cooperation remains to be elucidated. These findings uncover an epigenetic program in the control of serine and glycine synthesis in neuroblastoma cells [81][24].
The histone lysine methylation state is controlled not only by methyltransferases but also by demethylases. Members of the histone lysine demethylase family 4 (KDM4A-D) can remove di- (me2) and tri-methyl (me3) groups from H3K9 [106,107,108][49][50][51]. Thus, activation of the KDM4 family members could increase H3K9me1 levels by removing the repressive markers H3K9me2 and H3K9me3, thereby activating gene expression [108][51]. Indeed, it was found that KDM4C can epigenetically activate serine-glycine pathway genes under steady-state and serine deprivation conditions by removing the repressive marker H3K9me3. Again, this action of KDM4C requires ATF4. KDM4C activates ATF4 transcription and, in turn, ATF4 protein interacts with KDM4C to recruit it to the promoters of serine-glycine pathway genes for transcriptional activation [79][22].
Glycine is the end product in the serine-glycine synthesis pathway. Increased activation of this pathway could lead to the accumulation of glycine, which is detrimental to cells [109][52]. Glycine can be converted to the toxic metabolites aminoacetone and methylglyoxal [110][53]. In addition, glycine accumulation can reduce the flux of glucose carbon into the serine-glycine synthesis pathway [110,111][53][54]. The glycine cleavage system (GCS) has a major role in glycine breakdown in the mitochondria, which generates CO2, NH3, NADH, and the one-carbon unit 5,10-MTHF [112,113][55][56]. The GCS is composed of glycine decarboxylase (GLDC), aminomethyltransferase (AMT), dihydrolipoamide dehydrogenase (DLD), and glycine cleavage system protein H (GCSH). The first and rate-limiting step in GCS-mediated glycine breakdown is catalyzed by GLDC. The importance of the GCS in the maintenance of glycine homeostasis is evidenced by genetic findings that in mice and humans, GLDC mutations cause glycine encephalopathy (also known as non-ketotic hyperglycinemia) and neural tube defects, as a result of glycine accumulation [114,115,116][57][58][59].
Alptekin et al. reported that GLDC and serine-glycine pathway genes are co-upregulated in MYCN-amplified neuroblastoma tumors and cell lines. It was further shown that GLDC is a direct transcriptional target gene of MYCN. Depletion of GLDC by RNA interference disrupts multiple metabolic processes, affecting the TCA cycle and production of amino acids, purine nucleotides, and lipids. As a result, GLDC knockdown markedly inhibits the proliferation and tumorigenicity of MYCN-amplified neuroblastoma cell lines. These findings suggest a key role for glycine clearance in driving central carbon metabolism and biosynthesis of nucleotides and lipids in neuroblastoma cells [111][54].
In summary, the presented studies suggest that MYCN and ATF4 coordinate with the histone H3 methyltransferase G9A and the demethylase KDM4C to transcriptionally activate the serine-glycine biosynthesis pathway for increasing the production of serine, glycine, and one-carbon units in neuroblastoma cells.
2.3. Cysteine Metabolism
Cysteine is a sulfur-containing amino acid. Aside from being a proteinogenic amino acid, cysteine and its catabolic products are involved in many important cellular processes. Cysteine provides a highly reactive thiol side chain that functions as a nucleophile critical for the catalytic activity of many enzymes. The formation of disulfide bonds between cysteines is crucial for protein folding and stability. Further, cysteine is one of the three amino acid components of glutathione that counteracts reactive oxygen species (ROS) [117][60]. Moreover, cysteine catabolism produces hydrogen sulfide (H2S) to feed the electron transport chain for ATP production and pyruvate to supply carbon to the TCA cycle [118][61].
Cysteine can be synthesized de novo via the transsulfuration pathway, in which the enzyme cystathionine b-synthase (CBS) catalyzes the condensation of the methionine cycle intermediate homocysteine with serine to form cystathionine, which is then hydrolyzed by cystathionine γ-lyase (CTH) to generate cysteine and α-ketoglutarate. However, the major source of intracellular cysteine is the import of extracellular cystine coupled to the efflux of intracellular glutamate via the system Xc− transporter [119][62], which is composed of xCT (SLC7A11) that confers cystine transport function and 4F2hc (also known as SLC3A2) that serves as a chaperone to recruit SLC7A11 to the plasma membrane [73,120][16][63]. Once in the cytosol, cystine is readily reduced to produce two molecules of cysteine.
The expression of CBS, CTH, SLC3A2, and SLC7A11 is transcriptionally upregulated by KDM4C and ATF4 in neuroblastoma cells, leading to a significant increase in the intracellular levels of cysteine [79][22]. A recent study also reported that MYCN transcriptionally upregulates the system Xc− transporter to meet the increased demand of MYCN-amplified neuroblastoma cells to detoxify ROS by glutathione. This dependency creates a vulnerability that can be targeted by ferroptosis inducers [121][64]. Along the same lines, a more recent study showed that MYCN has a key role in maintaining cellular cysteine pools and that high MYCN expression sensitizes neuroblastoma cells to cystine deprivation, leading to lipid peroxidation and ferroptosis [122][65]. Together, these recent studies uncover an MYCN-dependent vulnerability to ferroptosis induction.
3. Reprogramming of Nucleotide Metabolism in Neuroblastoma
3.1. Purine Metabolism
In mammalian cells, de novo purine nucleotide synthesis is catalyzed by six enzymes (Figure 4), including phosphoribosyl pyrophosphate amidotransferase (PPAT), the trifunctional enzyme glycinamide ribonucleotide synthetase-aminoimidazole ribonucleotide synthetase-glycinamide ribonucleotide transformylase (GART), phosphoribosyl formylglycinamidine synthase (PFAS), and the bifunctional enzymes phosphoribosylaminoimidazole carboxylase (PAICS) and AICAR transformylase-IMP cyclohydrolase (ATIC) [124,125][66][67]. The starting substrate is 5-phosphoribosyl-1-pyrophosphate (PRPP), which is synthesized by PRPP synthase (PRPS1 and PRPS2) using ATP and ribose 5-phosphate generated by the pentose phosphate pathway. PPAT catalyzes the first and rate-limiting reaction that converts PRPP to 5-phosphoribosylamine (PRA). The end product of de novo purine synthesis is IMP, which is the precursor for all purine nucleotides. The purine base in IMP is composed of four nitrogen atoms (two from glutamine, one from aspartate, and one from glycine) and five carbons (two from glycine, two from the one-carbon carrier 10-formyl-THF, and one from CO2). The salvage pathway uses free purine nucleobases in the form of adenine, guanine, and hypoxanthine. These free bases are attached to PRPP by adenine phosphoribosyltransferase (APRT) to generate AMP or by hypoxanthine-guanine phosphoribosyltransferase (HPRT), which acts on hypoxanthine to form IMP and on guanine to form GMP [126][68].
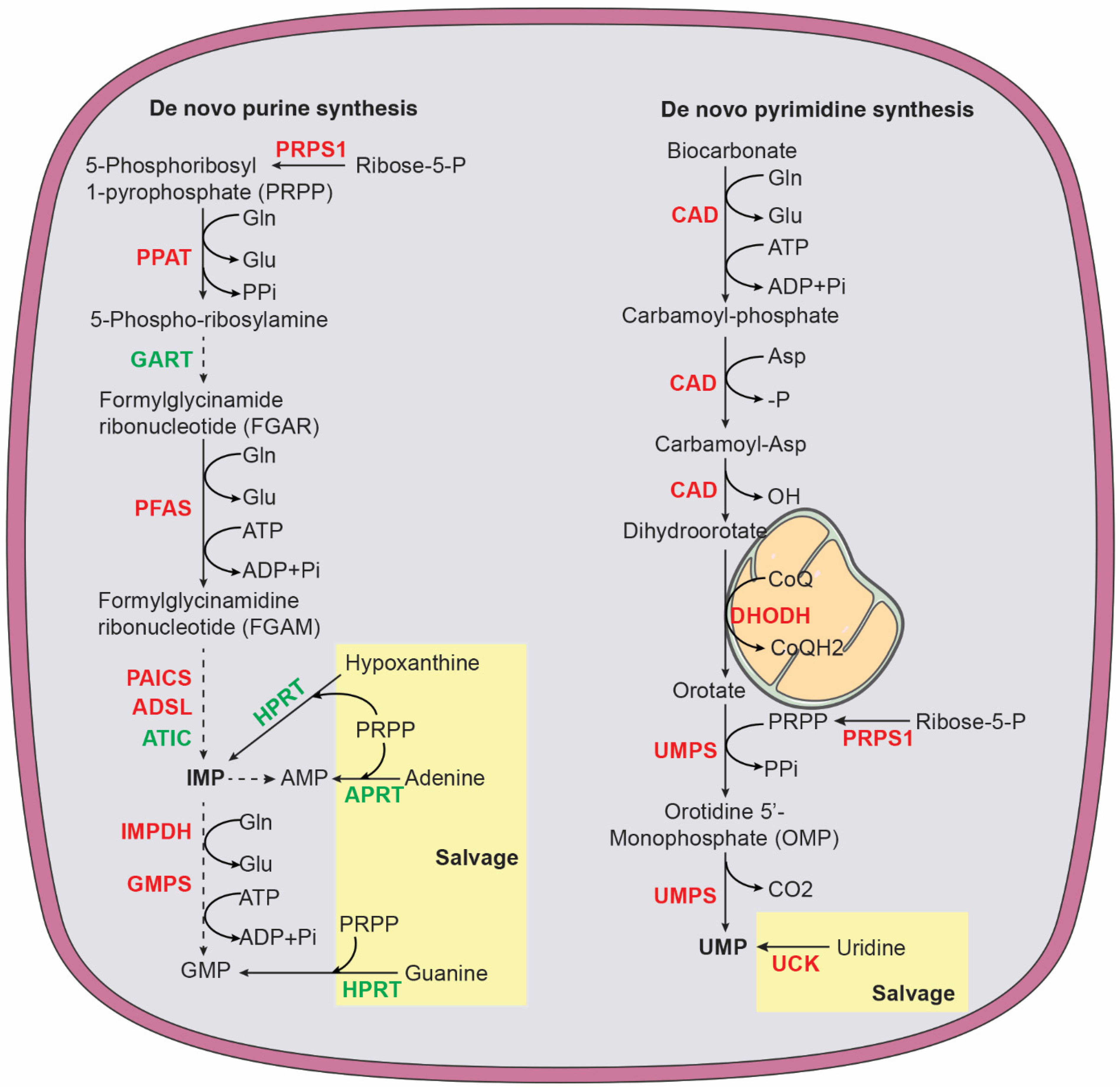
Figure 4. Purine and pyrimidine biosynthesis via de novo and salvage pathways. Purine pathway: ADSL, adenylosuccinate lyase; APRT, adenine phosphoribosyltransferase; ATIC, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase and IMP cyclohydrolase; GART, phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, phosphoribosylaminoimidazole synthetase; Glu, glutamate; Gln, glutamine; HPRT, hypoxanthine-guanine phosphoribosyltransferase; IMPDH, inosine monophosphate dehydrogenase; PRPS1, phosphoribosyl pyrophosphate synthetase 1; PAICS, phosphoribosyl aminoimidazole carboxylase and phosphoribosyl aminoimidazole succinocarboxamide synthase; PFAS, phosphoribosyl formylglycinamidine synthase; PPAT, phosphoribosyl pyrophosphate amidotransferase. Pyrimidine pathway: CAD, carbamoyl phosphate synthase, aspartate transcarbamoylase, and dihydroorotase; CoQ ubiquinone; CoQH2, ubiquinol; DHODH, dihydroorotate dehydrogenase; UCK, uridine-cytidine kinase; UMPS, UMP synthetase. Enzymes highlighted in red are MYCN targets.
MYCN has a major role in promoting purine biosynthesis in neuroblastoma cells. MYCN increases the production of glycine and 10-formyl-THF by transcriptionally upregulating enzymes in the serine-glycine-one-carbon network, with contributions from ATF4, KDM4C, and G9A [79,80,81][22][23][24]. MYCN also increases mRNA expression of enzymes involved in de novo purine synthesis, including PRPS1, PPAT, PFAS, PAICS, ADSL, and IMPDH1 [96][39]. In addition, chromatin immunoprecipitation and sequencing using anti-MYCN antibody provided evidence that MYCN directly targets PAICS and MTHFD2 for transcriptional upregulation [127][69]. MTHFD2 catalyzes the conversion of 5,10-MTHF to 10-formy-THF for donating carbon to purine ring synthesis (Figure 3).
3.2. Pyrimidine Metabolism
The primary function of pyrimidine nucleotides is to serve as building blocks for RNA and DNA synthesis. In addition, pyrimidine nucleotides have a key role in carbohydrate and lipid metabolism. Uridine diphosphate (UDP) sugars are substrates for all glycosylation reactions and glycogen synthesis and cytidine diphosphate (CDP)-diacylglycerol is required for the biosynthesis of complex glycerolipids [128,129,130][70][71][72]. In humans, pyrimidine nucleotides are produced by a combination of de novo biosynthesis and salvage (Figure 4). The trifunctional enzyme, composed of carbamoyl phosphate synthase, aspartate transcarbamoylase, and dihydroorotase (CAD), catalyzes the first step in de novo biosynthesis that generates dihydroorotate from glutamine, aspartate, and bicarbonate. Dihydroorotate is then oxidized to orotate by dihydroorotate dehydrogenase (DHODH), a mitochondrial membrane protein. This reaction is coupled to a reduction in ubiquinone (coenzyme Q, CoQ) to ubiquinol (CoQH2), linking pyrimidine nucleotide production to mitochondrial electron transport [131][73]. Next, the bifunctional enzyme UMP synthetase (UMPS) catalyzes the reaction that converts orotate into uridine monophosphate (UMP), which, in turn, serves as the precursor to generate all other pyrimidine nucleotides for RNA and DNA synthesis and carbohydrate and lipid metabolism. The salvage pathway recycles UMP and cytidine monophosphate (CMP) derived from intracellular RNA degradation or imports nucleosides (uridine and cytidine) from the bloodstream. Uridine-cytidine kinase (UCK) then converts uridine and cytidine into UMP and CMP, respectively. The pyrimidine ring is composed of four carbons (three from aspartate and one from bicarbonate) and two nitrogen atoms (one from glutamine and one from aspartate) [128,129][70][71].
MYCN coordinates multiple metabolic pathways to promote pyrimidine nucleotide synthesis. It activates de novo synthesis by transcriptional upregulation of CAD, DHODH, and UMPS [96][39]. It also cooperates with ATF4 to increase substrates for de novo synthesis by enhancing glutamine transport via transcriptional upregulation of SLC1A5 [93][36] and by increasing aspartate synthesis via upregulation of GOT1 and GOT2 [80,94][23][37]. In addition, MYCN increases UCK2 expression to activate the salvage pathway [96][39]. Moreover, MYCN transcriptionally upregulates the enzymes for dTMP synthesis, including thymidylate synthase and SHMT2 for producing 5,10-MTHF [80,96][23][39].
Collectively, the reported findings reveal an essential role for MYCN in enhancing nucleotide synthesis by transcriptional activation of synthesis enzymes and other metabolic pathways that provide substrates for nucleotide production. As a result, MYCN overexpression significantly increases intracellular pools of nucleotides in neuroblastoma cells [96][39].
References
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270.
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033.
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129.
- Shulkin, B.L.; Mitchell, D.S.; Ungar, D.R.; Prakash, D.; Dole, M.G.; Castle, V.P.; Hernandez, R.J.; Koeppe, R.A.; Hutchinson, R.J. Neoplasms in a pediatric population: 2--fluoro-2-deoxy-D-glucose PET studies. Radiology 1995, 194, 495–500.
- Levy, A.G.; Zage, P.E.; Akers, L.J.; Ghisoli, M.L.; Chen, Z.; Fang, W.; Kannan, S.; Graham, T.; Zeng, L.; Franklin, A.R.; et al. The combination of the novel glycolysis inhibitor 3-BrOP and rapamycin is effective against neuroblastoma. Investig. New Drugs 2012, 30, 191–199.
- Qing, G.; Skuli, N.; Mayes, P.A.; Pawel, B.; Martinez, D.; Maris, J.M.; Simon, M.C. Combinatorial Regulation of Neuroblastoma Tumor Progression by N-Myc and Hypoxia Inducible Factor HIF-1α. Cancer Res. 2010, 70, 10351–10361.
- Oliynyk, G.; Ruiz-Perez, M.V.; Sainero-Alcolado, L.; Dzieran, J.; Zirath, H.; Gallart-Ayala, H.; Wheelock, C.E.; Johansson, H.J.; Nilsson, R.; Lehtio, J.; et al. MYCN-enhanced Oxidative and Glycolytic Metabolism Reveals Vulnerabilities for Targeting Neuroblastoma. iScience 2019, 21, 188–204.
- Tjaden, B.; Baum, K.; Marquardt, V.; Simon, M.; Trajkovic-Arsic, M.; Kouril, T.; Siebers, B.; Lisec, J.; Siveke, J.T.; Schulte, J.H.; et al. N-Myc-induced metabolic rewiring creates novel therapeutic vulnerabilities in neuroblastoma. Sci. Rep. 2020, 10, 7157.
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039.
- Li, H.; Yang, F.; Hu, A.; Wang, X.; Fang, E.; Chen, Y.; Li, D.; Song, H.; Wang, J.; Guo, Y.; et al. Therapeutic targeting of circ-CUX1/EWSR1/MAZ axis inhibits glycolysis and neuroblastoma progression. EMBO Mol. Med. 2019, 11, e10835.
- Song, H.; Li, D.; Wang, X.; Fang, E.; Yang, F.; Hu, A.; Wang, J.; Guo, Y.; Liu, Y.; Li, H.; et al. HNF4A-AS1/hnRNPU/CTCF axis as a therapeutic target for aerobic glycolysis and neuroblastoma progression. J. Hematol. Oncol. 2020, 13, 24.
- Markert, C.L. Lactate dehydrogenase. Biochemistry and function of lactate dehydrogenase. Cell Biochem. Funct. 1984, 2, 131–134.
- Storey, K.B. Comparative enzymology—New insights from studies of an “old” enzyme, lactate dehydrogenase. Comp. Biochem. Physiol. Part. B Biochem. Mol. Biol. 2016, 199, 13–20.
- Dorneburg, C.; Fischer, M.; Barth, T.F.E.; Mueller-Klieser, W.; Hero, B.; Gecht, J.; Carter, D.R.; de Preter, K.; Mayer, B.; Christner, L.; et al. LDHA in Neuroblastoma Is Associated with Poor Outcome and Its Depletion Decreases Neuroblastoma Growth Independent of Aerobic Glycolysis. Clin. Cancer Res. 2018, 24, 5772–5783.
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid Transporters in Cancer and Their Relevance to “Glutamine Addiction”: Novel Targets for the Design of a New Class of Anticancer Drugs. Cancer Res. 2015, 75, 1782–1788.
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832.
- Choi, B.-H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675.
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443.
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21.
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395.
- Zhao, E.; Ding, J.; Xia, Y.; Liu, M.; Ye, B.; Choi, J.H.; Yan, C.; Dong, Z.; Huang, S.; Zha, Y.; et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. 2016, 14, 506–519.
- Xia, Y.; Ye, B.; Ding, J.; Yu, Y.; Alptekin, A.; Thangaraju, M.; Prasad, P.D.; Ding, Z.C.; Park, E.J.; Choi, J.H.; et al. Metabolic Reprogramming by MYCN Confers Dependence on the Serine-Glycine-One-Carbon Biosynthetic Pathway. Cancer Res. 2019, 79, 3837–3850.
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907.
- Liu, M.; Xia, Y.; Ding, J.; Ye, B.; Zhao, E.; Choi, J.H.; Alptekin, A.; Yan, C.; Dong, Z.; Huang, S.; et al. Transcriptional Profiling Reveals a Common Metabolic Program in High-Risk Human Neuroblastoma and Mouse Neuroblastoma Sphere-Forming Cells. Cell Rep. 2016, 17, 609–623.
- Eagle, H.; Oyama, V.I.; Levy, M.; Horton, C.L.; Fleischman, R. The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J. Biol. Chem. 1956, 218, 607–616.
- Kovacevic, Z.; McGivan, J.D. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol. Rev. 1983, 63, 547–605.
- Baggetto, L.G. Deviant energetic metabolism of glycolytic cancer cells. Biochimie 1992, 74, 959–974.
- Souba, W.W. Glutamine and cancer. Ann. Surg. 1993, 218, 715–728.
- Smith, R.J.; Wilmore, D.W. Glutamine nutrition and requirements. J. Parenter. Enter. Nutr. 1990, 14, 94S–99S.
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324.
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194.
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315.
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634.
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644.
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 2015, 235, 90–100.
- Wang, T.; Liu, L.; Chen, X.; Shen, Y.; Lian, G.; Shah, N.; Davidoff, A.M.; Yang, J.; Wang, R. MYCN drives glutaminolysis in neuroblastoma and confers sensitivity to an ROS augmenting agent. Cell Death Dis. 2018, 9, 220.
- Xiao, D.; Ren, P.; Su, H.; Yue, M.; Xiu, R.; Hu, Y.; Liu, H.; Qing, G. Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 2015, 6, 40655–40666.
- Yu, Y.; Ding, J.; Zhu, S.; Alptekin, A.; Dong, Z.; Yan, C.; Zha, Y.; Ding, H.-F. Therapeutic targeting of both dihydroorotate dehydrogenase and nucleoside transport in MYCN-amplified neuroblastoma. Cell Death Dis. 2021, 12, 821.
- Lockart, R.Z., Jr.; Eagle, H. Requirements for growth of single human cells. Science 1959, 129, 252–254.
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662.
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583.
- Reina-Campos, M.; Diaz-Meco, M.T.; Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 2019, 219, e201907022.
- Geeraerts, S.L.; Heylen, E.; De Keersmaecker, K.; Kampen, K.R. The ins and outs of serine and glycine metabolism in cancer. Nat. Metab. 2021, 3, 131–141.
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011, 25, 781–788.
- Peters, A.H.F.M.; Kubicek, S.; Mechtler, K.; O’Sullivan, R.J.; Derijck, A.A.H.A.; Perez-Burgos, L.; Kohlmaier, A.; Opravil, S.; Tachibana, M.; Shinkai, Y.; et al. Partitioning and Plasticity of Repressive Histone Methylation States in Mammalian Chromatin. Mol. Cell 2003, 12, 1577–1589.
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol. Cell 2003, 12, 1591–1598.
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791.
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179.
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507.
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 Histone Demethylases: Epigenetic Regulators in Cancer Cells. Cancer Res. 2013, 73, 2936–2942.
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258.
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367.
- Alptekin, A.; Ye, B.; Yu, Y.; Poole, C.J.; van Riggelen, J.; Zha, Y.; Ding, H.F. Glycine decarboxylase is a transcriptional target of MYCN required for neuroblastoma cell proliferation and tumorigenicity. Oncogene 2019, 38, 7504–7520.
- Wang, W.; Wu, Z.; Dai, Z.; Yang, Y.; Wang, J.; Wu, G. Glycine metabolism in animals and humans: Implications for nutrition and health. Amino Acids 2013, 45, 463–477.
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81.
- Narisawa, A.; Komatsuzaki, S.; Kikuchi, A.; Niihori, T.; Aoki, Y.; Fujiwara, K.; Tanemura, M.; Hata, A.; Suzuki, Y.; Relton, C.L.; et al. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum. Mol. Genet. 2012, 21, 1496–1503.
- Conter, C.; Rolland, M.O.; Cheillan, D.; Bonnet, V.; Maire, I.; Froissart, R. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J. Inherit. Metab. Dis. 2006, 29, 135–142.
- Pai, Y.J.; Leung, K.Y.; Savery, D.; Hutchin, T.; Prunty, H.; Heales, S.; Brosnan, M.E.; Brosnan, J.T.; Copp, A.J.; Greene, N.D. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat. Commun. 2015, 6, 6388.
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222.
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879.
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263.
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458.
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908.
- Alborzinia, H.; Florez, A.F.; Kreth, S.; Bruckner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat. Cancer 2022, 3, 471–485.
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154.
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485.
- Hartman, S.C.; Buchanan, J.M. The Biosynthesis of the Purines. In Ergebnisse der Physiologie Biologischen Chemie und Experimentellen Pharmakologie. Ergebnisse der Physiologie, Biologischen Chemie und Experimentellen Pharmakologie; Springer: Berlin/Heidelberg, Germany, 1959; Volume 50, pp. 75–121.
- Cheung, C.H.Y.; Hsu, C.L.; Tsuei, C.Y.; Kuo, T.T.; Huang, C.T.; Hsu, W.M.; Chung, Y.H.; Wu, H.Y.; Hsu, C.C.; Huang, H.C.; et al. Combinatorial targeting of MTHFD2 and PAICS in purine synthesis as a novel therapeutic strategy. Cell Death Dis. 2019, 10, 786.
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038.
- Okesli, A.; Khosla, C.; Bassik, M.C. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr. Opin. Biotechnol. 2017, 48, 127–134.
- Jennings, W.; Epand, R.M. CDP-diacylglycerol, a critical intermediate in lipid metabolism. Chem. Phys. Lipids 2020, 230, 104914.
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352.
More