Given the relevance of mitochondria in numerous physiological and pathological mechanisms, such as adenosine triphosphate (ATP) synthesis, apoptosis, metabolism, cancer progression, and drug resistance, mitochondrial genome (mtDNA) analysis has become of great interest in the study of human diseases, including cancer. To date, aA high number of variants and mutations have been identified in different types of tumors, which coexist with normal alleles, a phenomenon named heteroplasmy. This mechanism is considered an intermediate state between the fixation or elimination of the acquired mutations. It is suggested that mutations, which confer adaptive advantages to tumor growth and invasion, are enriched in malignant cells.
- heteroplasmy
- heteroplasmy shifting
- mitochondrial DNA
- mitochondrial mutations
- cancer
1. Introduction
2. Mitochondrial Genome
|
Gene Symbol |
Name |
Function |
OXPHOS Complex |
Nucleotide Start Position |
Nucleotide End Position |
|---|---|---|---|---|---|
|
MT-ATP6 |
ATP synthase membrane subunit 6 |
Proton transmembrane transporter activity |
V |
8527 |
9207 |
|
MT-ATP8 |
ATP synthase membrane subunit 8 |
Proton transmembrane transporter activity |
8366 |
8572 |
|
|
MT-CO1 |
Cytochrome c oxidase I |
Electron transport Heme binding |
5904 |
7445 |
|
|
MT-CO2 |
Cytochrome c oxidase II |
Electron transport |
IV |
7586 |
8269 |
|
MT-CO3 |
Cytochrome c oxidase III |
Electron transport |
9207 |
9990 |
|
|
MT-CYB |
Cytochrome b |
Electron transport Heme binding |
III |
14747 |
15887 |
|
MT-ND1 |
NADH: ubiquinone oxidoreductase core subunit 1 |
Proton pumping Reduction site for ubiquinone |
3307 |
4262 |
|
|
MT-ND2 |
NADH: ubiquinone oxidoreductase core subunit 2 |
Proton pumping |
4470 |
5511 |
|
|
MT-ND3 |
NADH: ubiquinone oxidoreductase core subunit 3 |
Proton pumping |
I |
10059 |
10404 |
|
MT-ND4 |
NADH: ubiquinone oxidoreductase core subunit 4 |
Proton pumping |
10760 |
12137 |
|
|
MT-ND4L |
NADH: ubiquinone oxidoreductase core subunit 4L |
Proton pumping |
10470 |
10766 |
|
|
MT-ND5 |
NADH:ubiquinone oxidoreductase core subunit 5 |
Proton pumping |
12337 |
14148 |
|
|
MT-ND6 |
NADH:ubiquinone oxidoreductase core subunit 6 |
Proton pumping |
14149 |
14673 |
|
|
MT-RNR1 |
12S rRNA |
12S ribosomal RNA Small subunit |
- |
648 |
1601 |
|
MT-RNR2 |
16S rRNA |
16S ribosomal RNA Large subunit |
- |
1671 |
3229 |
|
MT-TA |
tRNA-Ala |
tRNA for alanine |
- |
5587 |
5655 |
|
MT-TC |
tRNA-Cys |
tRNA for cysteine |
- |
5761 |
5826 |
|
MT-TD |
tRNA-Asp |
tRNA for aspartic acid |
- |
7518 |
7585 |
|
MT-TE |
tRNA-Glu |
tRNA for glutamic acid |
- |
14674 |
14742 |
|
MT-TF |
tRNA-Phe |
tRNA for phenylalanine |
- |
577 |
647 |
|
MT-TG |
tRNA-Gly |
tRNA for glycine |
- |
9991 |
10058 |
|
MT-TH |
tRNA-His |
tRNA for histidine |
- |
12138 |
12206 |
|
MT-TI |
tRNA-Ile |
tRNA for isoleucine |
- |
4263 |
4331 |
|
MT-TK |
tRNA-Lys |
tRNA for lysine |
- |
8295 |
8364 |
|
MT-TL1 |
tRNA-Leu (UUA/G) 1 |
tRNA for leucine 1 |
- |
3230 |
3304 |
|
MT-TL2 |
tRNA-Leu (CUN) 2 |
tRNA for leucine 2 |
- |
12266 |
12336 |
|
MT-TM |
tRNA-Met |
tRNA for methionine |
- |
4402 |
4469 |
|
MT-TN |
tRNA-Asn |
tRNA for asparagine |
- |
5657 |
5729 |
|
MT-TP |
tRNA-Pro |
tRNA for proline |
- |
15956 |
16023 |
|
MT-TQ |
tRNA-Gln |
tRNA for glutamine |
- |
4329 |
4400 |
|
MT-TR |
tRNA-Arg |
tRNA for arginine |
- |
10405 |
10469 |
|
MT-TS1 |
tRNA-Ser (UCN) 1 |
tRNA for serine 1 |
- |
7446 |
7514 |
|
MT-TS2 |
tRNA-Ser (AGU/C) 2 |
tRNA for serine 2 |
- |
12207 |
12265 |
|
MT-TT |
tRNA-Thr |
tRNA for threonine |
- |
15888 |
15953 |
|
MT-TV |
tRNA-Val |
tRNA for valine |
- |
1602 |
1670 |
|
MT-TW |
tRNA-Trp |
tRNA for tryptophan |
- |
5512 |
5579 |
|
MT-TY |
tRNA-Tyr |
tRNA for tyrosine |
- |
5826 |
5891 |
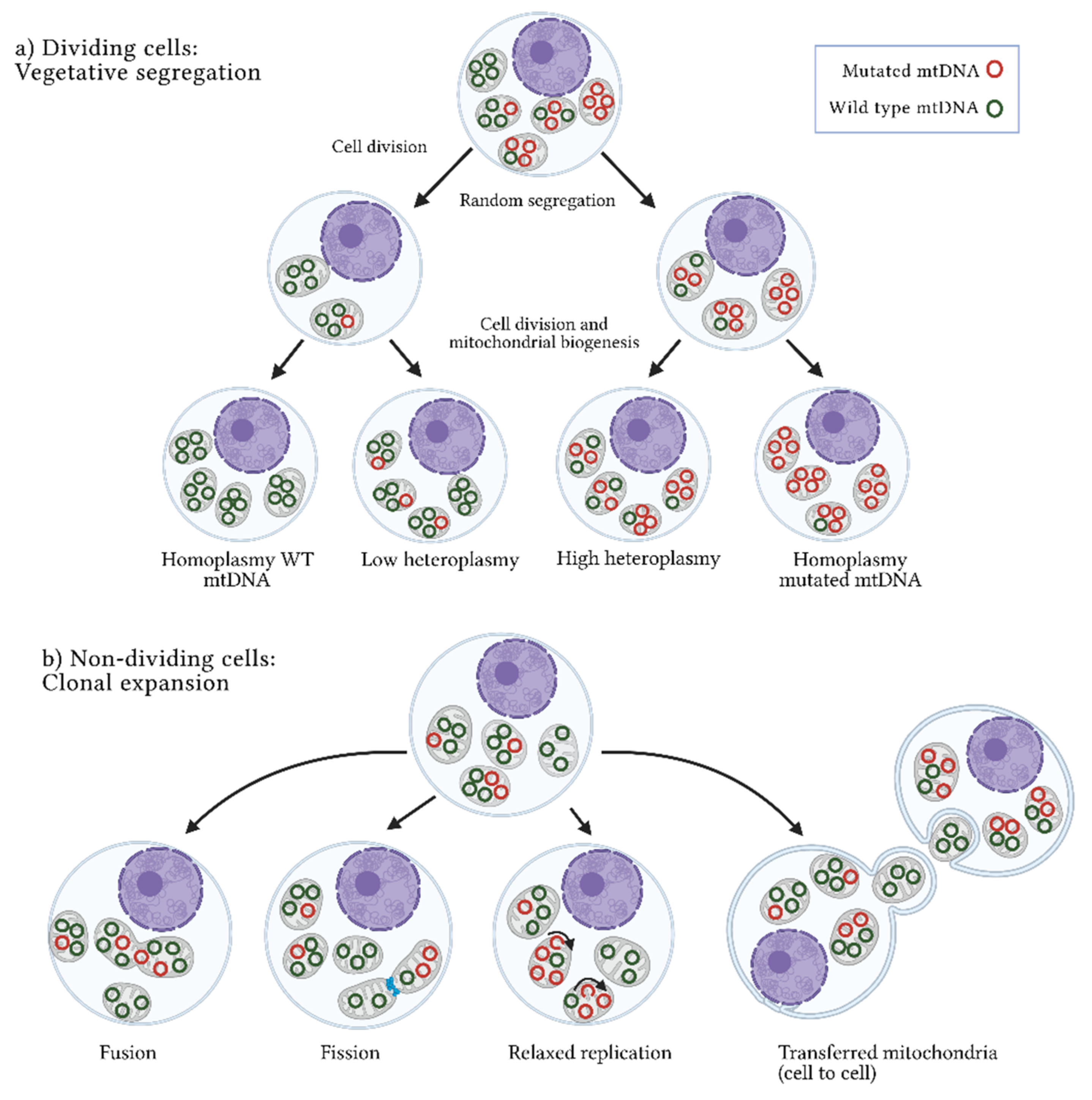
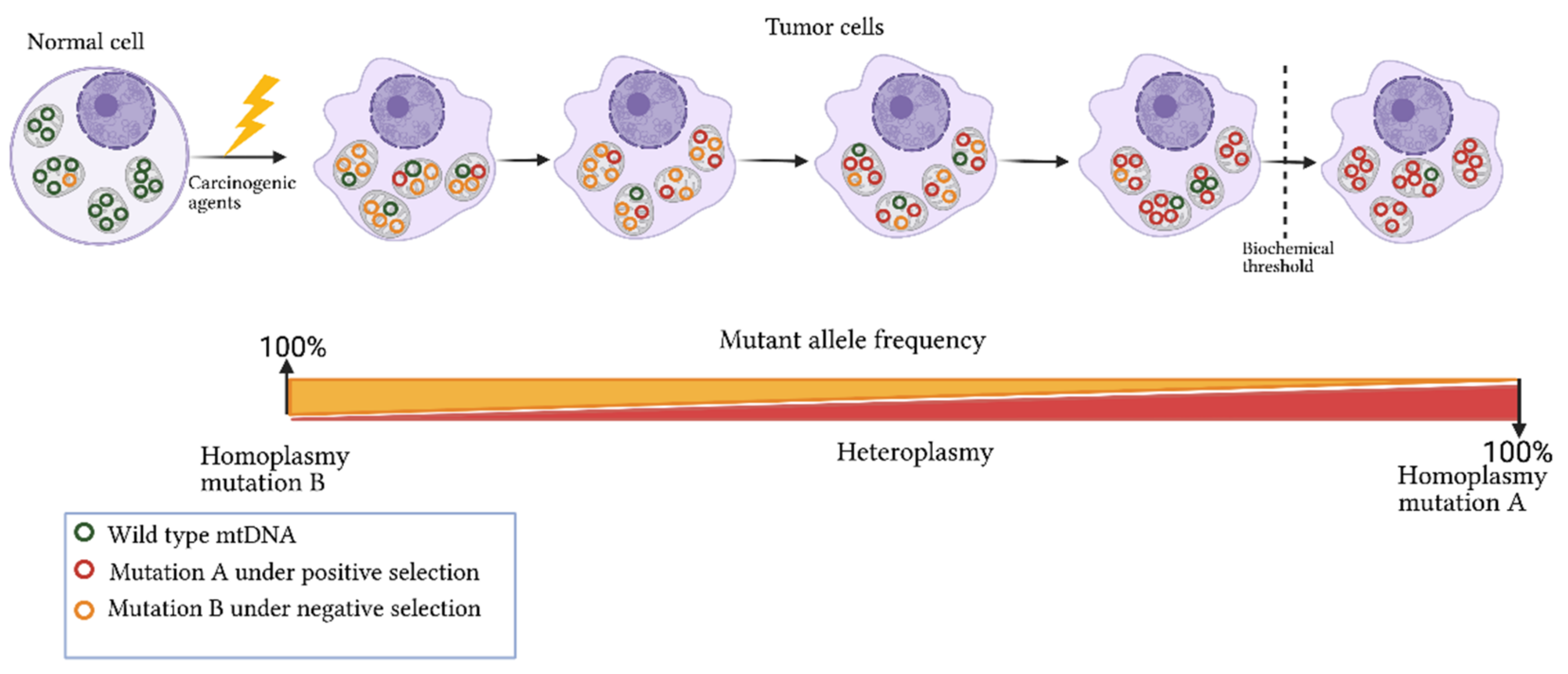
3. Origins of Heteroplasmy and Its Impact on Disease
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res. 2018, 78, 631–644.
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421.
- Zhang, Z.; Yang, L.; Li, Y.; Wu, Y.; Li, X.; Wu, X. Four long noncoding RNAs act as biomarkers in lung adenocarcinoma. Open Med. 2021, 16, 660–671.
- Ghafarpour, V.; Khansari, M.; Banaei-Moghaddam, A.M.; Najafi, A.; Masoudi-Nejad, A. DNA methylation association with stage progression of head and neck squamous cell carcinoma. Comput. Biol. Med. 2021, 134, 104473.
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586.
- Hägg, S.; Jylhävä, J.; Wang, Y.; Czene, K.; Grassmann, F. Correction to: Deciphering the genetic and epidemiological landscape of mitochondrial DNA abundance. Qual. Life Res. 2021, 140, 863.
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020.
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769.
- Basu, S.; Xie, X.; Uhler, J.P.; Hedberg-Oldfors, C.; Milenkovic, D.; Baris, O.R.; Kimoloi, S.; Matic, S.; Stewart, J.B.; Larsson, N.-G.; et al. Accurate mapping of mitochondrial DNA deletions and duplications using deep sequencing. PLoS Genet. 2020, 16, e1009242.
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676.
- Rao, S.G. Mitochondrial Changes in Cancer. Organotypic Models Drug Dev. 2016, 240, 211–227.
- Bussard, K.M.; Siracusa, L.D. Understanding Mitochondrial Polymorphisms in Cancer. Cancer Res. 2017, 77, 6051–6059.
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352.
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martin, S.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, 3.
- Li, L.; Chen, L.; Li, J.; Zhang, W.; Liao, Y.; Chen, J.; Sun, Z. Correlational study on mitochondrial DNA mutations as potential risk factors in breast cancer. Oncotarget 2016, 7, 31270–31283.
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic shifts in tumor mitochondrial genomes reveal tissue-specific signals of relaxed and positive selection. Hum. Mol. Genet. 2017, 26, 2912–2922.
- Hahn, A.; Zuryn, S. The Cellular Mitochondrial Genome Landscape in Disease. Trends Cell Biol. 2019, 29, 227–240.
- Tikochinski, Y.; Carreras, C.; Tikochinski, G.; Vilaca, S.T. Population-specific signatures of intra-individual mitochondrial DNA heteroplasmy and their potential evolutionary advantages. Sci. Rep. 2020, 10, 211.
- Qi, Y.; Wei, Y.; Wang, Q.; Xu, H.; Wang, Y.; Yao, A.; Yang, H.; Gao, Y.; Zhou, F. Heteroplasmy of mutant mitochondrial DNA A10398G and analysis of its prognostic value in non-small cell lung cancer. Oncol. Lett. 2016, 12, 3081–3088.
- Fendt, L.; Fazzini, F.; Weissensteiner, H.; Bruckmoser, E.; Schönherr, S.; Schäfer, G.; Losso, J.L.; Streiter, G.A.; Lamina, C.; Rasse, M.; et al. Profiling of Mitochondrial DNA Heteroplasmy in a Prospective Oral Squamous Cell Carcinoma Study. Cancers 2020, 12, 1933.
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054.
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 1–8.
- Kloss-Brandstätter, A.; Schäfer, G.; Erhart, G.; Hüttenhofer, A.; Coassin, S.; Seifarth, C.; Summerer, M.; Bektic, J.; Klocker, H.; Kronenberg, F. Somatic Mutations throughout the Entire Mitochondrial Genome Are Associated with Elevated PSA Levels in Prostate Cancer Patients. Am. J. Hum. Genet. 2010, 87, 802–812.
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274.
- Margulis, L. Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp. Soc. Exp. Biol. 1975, 29, 21–38.
- Yamauchi, A. Rate of Gene Transfer From Mitochondria to Nucleus: Effects of Cytoplasmic Inheritance System and Intensity of Intracellular Competition. Genetics 2005, 171, 1387–1396.
- Anderson, S.E.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465.
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell. Longev. 2017, 2017, 1–15.
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662.
- Hou, X.-S.; Wang, H.-S.; Mugaka, B.P.; Yang, G.-J.; Ding, Y. Mitochondria: Promising organelle targets for cancer diagnosis and treatment. Biomater. Sci. 2018, 6, 2786–2797.
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223.
- O’Hara, R.; Tedone, E.; Ludlow, A.T.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative mitochondrial DNA copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 2019, 29, 1878–1888.
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21.
- Dong, J.; Wong, L.-J.; Mims, M.P. Mitochondrial inheritance and cancer. Transl. Res. 2018, 202, 24–34.
- Lajbner, Z.; Pnini, R.; Camus, M.F.; Miller, J.; Dowling, D. Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci. Rep. 2018, 8, 9500.
- Motoi, M.; Nishimura, T.; Egashira, Y.; Kishida, F.; Watanuki, S. Relationship between mitochondrial haplogroup and physiological responses to hypobaric hypoxia. J. Physiol. Anthr. 2016, 35, 12.
- Chen, Y.; Gong, L.; Liu, X.; Chen, X.; Yang, S.; Luo, Y. Mitochondrial DNA genomes revealed different patterns of high-altitude adaptation in high-altitude Tajiks compared with Tibetans and Sherpas. Sci. Rep. 2020, 10, 1–9.
- Toncheva, D.; Serbezov, D.; Karachanak-Yankova, S.; Nesheva, D. Ancient mitochondrial DNA pathogenic variants putatively associated with mitochondrial disease. PLoS ONE 2020, 15, e0233666.
- Xiao, F.; Li, M.; Wang, J.; Liu, J.; Li, J.; Fang, H.; Lyu, J.; Shen, L. Association between mitochondrial DNA haplogroup variation and coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 960–966.
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van De Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146.
- Ma, L.; Fu, Q.; Xu, B.; Zhou, H.; Gao, J.; Shao, X.; Xiong, J.; Gu, Q.; Wen, S.; Li, F.; et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int. J. Cancer 2018, 142, 1786–1796.
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70.
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664.
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. The second genome: Effects of the mitochondrial genome on cancer progression. Adv. Cancer Res. 2019, 142, 63–105.
- Wu, C.-W.; Yin, P.-H.; Hung, W.-Y.; Li, A.F.-Y.; Li, S.-H.; Chi, C.-W.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosom. Cancer 2005, 44, 19–28.
- Yadav, N.; Chandra, D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1836, 336–344.
- Wallace, D.C. Bioenergetic Origins of Complexity and Disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16.
- Stewart, J.B.; Chinnery, P.F. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 2021, 22, 106–118.
- Zheng, W.; Khrapko, K.; Coller, H.A.; Thilly, W.G.; Copeland, W.C. Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutat. Res. 2006, 599, 11–20.
- Wisnovsky, S.; Sack, T.; Pagliarini, D.J.; Laposa, R.R.; Kelley, S.O. DNA Polymerase theta Increases Mutational Rates in Mitochondrial DNA. ACS Chem. Biol. 2018, 13, 900–908.
- Gustafson, M.A.; Sullivan, E.D.; Copeland, W.C. Consequences of compromised mitochondrial genome integrity. DNA Repair 2020, 93, 102916.
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104.
- Jimenez-Morales, S.; Perez-Amado, C.J.; Langley, E.; Hidalgo-Miranda, A. Overview of mitochondrial germline variants and mutations in human disease: Focus on breast cancer (Review). Int. J. Oncol. 2018, 53, 923–936.
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. Adv. Exp. Med. Biol. 2017, 982, 577–594.
- Ahn, E.H.; Hirohata, K.; Kohrn, B.F.; Fox, E.J.; Chang, C.-C.; Loeb, L.A. Detection of Ultra-Rare Mitochondrial Mutations in Breast Stem Cells by Duplex Sequencing. PLoS ONE 2015, 10, e0136216.
- Zhang, H.; Burr, S.; Chinnery, P.F. The mitochondrial DNA genetic bottleneck: Inheritance and beyond. Essays Biochem. 2018, 62, 225–234.
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612.
- Floros, V.I.; Pyle, A.; Dietmann, S.; Wei, W.; Tang, W.C.W.; Irie, N.; Payne, B.; Capalbo, A.; Noli, L.; Coxhead, J.; et al. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 2018, 20, 144–151.
- Wei, W.; Chinnery, P.F. Inheritance of mitochondrial DNA in humans: Implications for rare and common diseases. J. Intern. Med. 2020, 287, 634–644.
- Johnston, I.; Burgstaller, J.P.; Havlicek, V.; Kolbe, T.; Rülicke, T.; Brem, G.; Poulton, J.; Jones, N.S. Stochastic modelling, Bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism. eLife 2015, 4, e07464.
- Naeem, M.M.; Sondheimer, N. Heteroplasmy Shifting as Therapy for Mitochondrial Disorders. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; pp. 257–267.
- Jackson, M.V.; Krasnodembskaya, A.D. Analysis of Mitochondrial Transfer in Direct Co-cultures of Human Monocyte-derived Macrophages (MDM) and Mesenchymal Stem Cells (MSC). Bio-Protocol 2017, 7, e2255.
- Brestoff, J.R.; Wilen, C.B.; Moley, J.R.; Li, Y.; Zou, W.; Malvin, N.P.; Rowen, M.N.; Saunders, B.T.; Ma, H.; Mack, M.R.; et al. Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell Metab. 2021, 33, 270–282.e8.
- Han, D.; Zheng, X.; Wang, X.; Jin, T.; Cui, L.; Chen, Z. Mesenchymal Stem/Stromal Cell-Mediated Mitochondrial Transfer and the Therapeutic Potential in Treatment of Neurological Diseases. Stem Cells Int. 2020, 2020, 1–16.
- She, Z.; Xie, M.; Hun, M.; Abdirahman, A.S.; Li, C.; Wu, F.; Luo, S.; Wan, W.; Wen, C.; Tian, J. Immunoregulatory Effects of Mitochondria Transferred by Extracellular Vesicles. Front. Immunol. 2021, 11, 628576.
- Burgstaller, J.P.; Kolbe, T.; Havlicek, V.; Hembach, S.; Poulton, J.; Piálek, J.; Steinborn, R.; Rülicke, T.; Brem, G.; Jones, N.S.; et al. Large-scale genetic analysis reveals mammalian mtDNA heteroplasmy dynamics and variance increase through lifetimes and generations. Nat. Commun. 2018, 9, 2488.
- Lechuga-Vieco, A.V.; Latorre-Pellicer, A.; Johnston, I.G.; Prota, G.; Gileadi, U.; Justo-Méndez, R.; Acín-Pérez, R.; Martínez-De-Mena, R.; Fernández-Toro, J.M.; Jimenez-Blasco, D.; et al. Cell identity and nucleo-mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Sci. Adv. 2020, 6, eaba5345.
- Zaidi, A.A.; Wilton, P.R.; Su, M.S.-W.; Paul, I.M.; Arbeithuber, B.; Anthony, K.; Nekrutenko, A.; Nielsen, R.; Makova, K.D. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc. Natl. Acad. Sci. USA 2019, 116, 25172–25178.
- Li, M.; Schröder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496.
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuziodonahue, C.A.; Markowitz, S.D.; Velculescu, V.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nat. Cell Biol. 2010, 464, 610–614.
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659.
- Zekonyte, U.; Bacman, S.R.; Moraes, C.T. DNA-editing enzymes as potential treatments for heteroplasmic mtDNA diseases. J. Intern. Med. 2020, 287, 685–697.
- Sacconi, S.; Salviati, L.; Nishigaki, Y.; Walker, W.F.; Hernandez-Rosa, E.; Trevisson, E.; Delplace, S.; Desnuelle, C.; Shanske, S.; Hirano, M.; et al. A functionally dominant mitochondrial DNA mutation. Hum. Mol. Genet. 2008, 17, 1814–1820.
- Yapa, N.M.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204.
- Ameele, J.V.D.; Li, A.Y.; Ma, H.; Chinnery, P.F. Mitochondrial heteroplasmy beyond the oocyte bottleneck. Semin. Cell Dev. Biol. 2020, 97, 156–166.
- Stefano, G.B.; Kream, R.M. Mitochondrial DNA heteroplasmy in human health and disease. Biomed. Rep. 2016, 4, 259–262.
- Kopinski, P.K.; Janssen, K.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.; Campbell, S.L.; et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035.
- McMillan, R.P.; Stewart, S.; Budnick, J.A.; Caswell, C.C.; Hulver, M.W.; Mukherjee, K.; Srivastava, S. Quantitative Variation in m.3243A > G Mutation Produce Discrete Changes in Energy Metabolism. Sci. Rep. 2019, 9, 1–11.