Given the relevance of mitochondria in numerous physiological and pathological mechanisms, such as adenosine triphosphate (ATP) synthesis, apoptosis, metabolism, cancer progression, and drug resistance, mitochondrial genome (mtDNA) analysis has become of great interest in the study of human diseases, including cancer. ATo date, a high number of variants and mutations have been identified in different types of tumors, which coexist with normal alleles, a phenomenon named heteroplasmy. This mechanism is considered an intermediate state between the fixation or elimination of the acquired mutations. It is suggested that mutations, which confer adaptive advantages to tumor growth and invasion, are enriched in malignant cells.
1. Introduction
Despite the growing understanding of cancer biology and the constant effort to develop more effective diagnostic, follow-up and therapeutic strategies, these diseases still have important socio–economic implications worldwide
[1]. To identify new diagnostic and prognostic biomarkers and even new potential molecular targets, current research focuses on the nuclear genome by detecting mutations and on gene expression and epigenomic signatures
[2][3][4][5][2,3,4,5]; however, given the number of biological processes in which mitochondria participate (energy synthesis, metabolism, apoptosis and signaling pathways, among others), these organelles are gaining great relevance in addressing these questions
[6]. More specifically, it has been shown that the mitochondrial DNA (mtDNA) of tumor cells accumulates many alterations, including punctual mutations, insertions, deletions, and variations in mtDNA copy number (mtCNV), which trigger the metabolic reprogramming of transformed cells as a biological strategy that favors tumor cell proliferation and survival
[7][8][9][10][11][12][13][7,8,9,10,11,12,13]. Even though the mutational spectrum of mtDNA has already been studied in different types of cancer, the mechanism by which mtDNA mutations contribute to tumor development is not yet well known
[14][15][16][14,15,16]. Additionally, heteroplasmy, defined as the presence of two or more mtDNA variants coexisting within the same cell, and the proportion of mutated mtDNA (the heteroplasmy level) are crucial for the expression of specific pathological phenotypes
[17][18][19][17,18,19]. Heteroplasmy has been proposed as an intermediate phase between the fixation or elimination of newly acquired mtDNA mutations. However, the critical threshold to observe a biochemical defect in the respiratory chain or for disease expression is undisclosed in cancer and scarcely analyzed in the studies that characterize the mtDNA variant landscape. Interestingly, many studies have shown an association among heteroplasmic levels and risk and survival of cancer
[20][21][22][23][24][20,21,22,23,24].
2. Mitochondrial Genome
The mitochondrion is a semi-autonomous organelle whose main function is the synthesis of cellular adenosine triphosphate (ATP). However, mitochondria also play roles in metabolism regulation, cell death, and signaling pathways involved in cell growth and proliferation
[6]. The endosymbiotic theory indicates that the mitochondrion originated from endocytosis of an alpha-protobacterium by an ancestral pre-eukaryotic cell
[6][25][26][6,25,26]. Throughout evolution, most of the mtDNA has migrated to the nucleus, leaving only a 16,569 base pair (bp) circular molecule
[27]. This genome is constituted by two circular chains, a heavy strand (H) and a light strand (L) enriched in guanine and cytosine, respectively. mtDNA has 37 genes; two transcribe for ribosomal RNA (rRNA), 22 transcribe for transfer RNAs (tRNAs) and 13 transcribe for gene-encoding protein subunits of the enzyme complex of the oxidative phosphorylation system (OXPHOS) (
Table 1). Additionally, the mitochondrial genome has a non-coding region of 1.1 kilobases (Kb), named D-Loop, which contains two hypervariable regions (HVR1 and HVR2) and sequences involved in the control of mtDNA replication and gene transcription
[28][29][28,29]. The lack of histones, efficient DNA repair mechanisms, and the proximity to reactive oxygen species (ROS) generated by the OXPHOS system (mainly from Complex I and III) explain the high mutation rate observed in mtDNA, which is between 10 and 17 times higher than that of the nuclear genome (nDNA)
[28][29][30][31][28,29,30,31]. Furthermore, mtDNA is a multicopy genome, displaying variable copy numbers over time, among cell types, tissues, subjects, physiological conditions. The estimated copy number in mammalians ranges from 100 to 10,000 molecules per cell depending on the cell type and its energy requirement
[32][33][34][32,33,34]. In humans, mtDNA is only inherited through the maternal line, and all alleles are passed down together as a single unit called haplotype, which could be shared in populations with common ancestry. In fact, a set of haplotypes or haplogroup is useful for identifying ethnic groups or populations
[35]. Studies suggest that specific haplogroups have conferred environmental adaptive advantages but are also associated with diseases such as cancer
[36][37][38][39][40][41][42][36,37,38,39,40,41,42].
Table 1.
Encoded mitochondrial DNA genes.
|
Gene
Symbol
|
Name
|
Function
|
OXPHOS Complex
|
Nucleotide Start Position
|
Nucleotide End Position
|
|
MT-ATP6
|
ATP synthase membrane subunit 6
|
Proton transmembrane transporter activity
|
V
|
8527
|
9207
|
|
MT-ATP8
|
ATP synthase membrane subunit 8
|
Proton transmembrane transporter activity
|
|
8366
|
8572
|
|
MT-CO1
|
Cytochrome c oxidase I
|
Electron transport
Heme binding
|
|
5904
|
7445
|
|
MT-CO2
|
Cytochrome c oxidase II
|
Electron transport
|
IV
|
7586
|
8269
|
|
MT-CO3
|
Cytochrome c oxidase III
|
Electron transport
|
|
9207
|
9990
|
|
MT-CYB
|
Cytochrome b
|
Electron transport
Heme binding
|
III
|
14747
|
15887
|
|
MT-ND1
|
NADH: ubiquinone oxidoreductase core subunit 1
|
Proton pumping
Reduction site for ubiquinone
|
|
3307
|
4262
|
|
MT-ND2
|
NADH: ubiquinone oxidoreductase core subunit 2
|
Proton pumping
|
|
4470
|
5511
|
|
MT-ND3
|
NADH: ubiquinone oxidoreductase core subunit 3
|
Proton pumping
|
I
|
10059
|
10404
|
|
MT-ND4
|
NADH: ubiquinone oxidoreductase core subunit 4
|
Proton pumping
|
|
10760
|
12137
|
|
MT-ND4L
|
NADH: ubiquinone oxidoreductase core subunit 4L
|
Proton pumping
|
|
10470
|
10766
|
|
MT-ND5
|
NADH:ubiquinone oxidoreductase core subunit 5
|
Proton pumping
|
|
12337
|
14148
|
|
MT-ND6
|
NADH:ubiquinone oxidoreductase core subunit 6
|
Proton pumping
|
|
14149
|
14673
|
|
MT-RNR1
|
12S rRNA
|
12S ribosomal RNA Small subunit
|
-
|
648
|
1601
|
|
MT-RNR2
|
16S rRNA
|
16S ribosomal RNA Large subunit
|
-
|
1671
|
3229
|
|
MT-TA
|
tRNA-Ala
|
tRNA for alanine
|
-
|
5587
|
5655
|
|
MT-TC
|
tRNA-Cys
|
tRNA for cysteine
|
-
|
5761
|
5826
|
|
MT-TD
|
tRNA-Asp
|
tRNA for aspartic acid
|
-
|
7518
|
7585
|
|
MT-TE
|
tRNA-Glu
|
tRNA for glutamic acid
|
-
|
14674
|
14742
|
|
MT-TF
|
tRNA-Phe
|
tRNA for phenylalanine
|
-
|
577
|
647
|
|
MT-TG
|
tRNA-Gly
|
tRNA for glycine
|
-
|
9991
|
10058
|
|
MT-TH
|
tRNA-His
|
tRNA for histidine
|
-
|
12138
|
12206
|
|
MT-TI
|
tRNA-Ile
|
tRNA for isoleucine
|
-
|
4263
|
4331
|
|
MT-TK
|
tRNA-Lys
|
tRNA for lysine
|
-
|
8295
|
8364
|
|
MT-TL1
|
tRNA-Leu (UUA/G) 1
|
tRNA for leucine 1
|
-
|
3230
|
3304
|
|
MT-TL2
|
tRNA-Leu (CUN) 2
|
tRNA for leucine 2
|
-
|
12266
|
12336
|
|
MT-TM
|
tRNA-Met
|
tRNA for methionine
|
-
|
4402
|
4469
|
|
MT-TN
|
tRNA-Asn
|
tRNA for asparagine
|
-
|
5657
|
5729
|
|
MT-TP
|
tRNA-Pro
|
tRNA for proline
|
-
|
15956
|
16023
|
|
MT-TQ
|
tRNA-Gln
|
tRNA for glutamine
|
-
|
4329
|
4400
|
|
MT-TR
|
tRNA-Arg
|
tRNA for arginine
|
-
|
10405
|
10469
|
|
MT-TS1
|
tRNA-Ser (UCN) 1
|
tRNA for serine 1
|
-
|
7446
|
7514
|
|
MT-TS2
|
tRNA-Ser (AGU/C) 2
|
tRNA for serine 2
|
-
|
12207
|
12265
|
|
MT-TT
|
tRNA-Thr
|
tRNA for threonine
|
-
|
15888
|
15953
|
|
MT-TV
|
tRNA-Val
|
tRNA for valine
|
-
|
1602
|
1670
|
|
MT-TW
|
tRNA-Trp
|
tRNA for tryptophan
|
-
|
5512
|
5579
|
|
MT-TY
|
tRNA-Tyr
|
tRNA for tyrosine
|
-
|
5826
|
5891
|
ATP: adenosine triphosphate; NADH: nicotinamide adenine dinucleotide reduced; OXPHOS: oxidative phosphorylation system, I: NADH: ubiquinone oxidoreductase complex; III: cytochrome bc1 complex; IV: Cytochrome c Oxidase complex; V: Mitochondrial ATP synthase complex; -: no OXPHOS complex.
Even though mtDNA encodes only 13 of the approximately 1500 proteins that conform to the mitochondria, its maintenance is essential for mitochondrial biogenesis
[6][43][6,43]. The presence of alterations in mtDNA correlates with an inefficient OXPHOS system, decreased ATP synthesis, and an increased ROS production rate, phenomena associated with pathological phenotypes
[44][45][46][47][48][44,45,46,47,48].
3. Origins of Heteroplasmy and Its Impact on Disease
mtDNA replication, mediated by DNA polymerase γ (DNApolγ), is continuous and cell-cycle-independent. Replication is the primary source of new mutations in mtDNA. Despite the high fidelity of DNApolγ, the elevated replication rate and limited DNA repair mechanisms contribute to the maintenance of acquired mutations (point mutations, insertions, deletions, etc.)
[10][49][50][51][52][53][10,49,50,51,52,53]. Due to the polyploid nature of mtDNA, mutations can be found in different proportions within a mitochondrion, cell, tissue, or individual, giving rise to heteroplasmy
[18][31][45][54][55][18,31,45,54,55]. Heteroplasmy can be higher if the mutated allele frequency (MAF) ranges from 20% to 95% or lower if the MAF ranges from 0.5% to 20%. A MAF <0.5% corresponds to a rare variant. If the allele represents more than 95% of the mtDNA molecules, this is considered homoplasmic
[54][56][54,56].
Heteroplasmy is maternally inherited, and its levels are determined during oogenesis. The proportion of mutated alleles differs between oocytes and then among offspring. Shifts in heteroplasmy levels have been observed in humans within a few generations, and a genetic bottleneck during the maternal transmission of mtDNA has been suggested to explain this phenomenon. The bottleneck could be derived from a reduction in the numbers of mtDNA molecules by three mechanisms: (a) passive reduction, where the mtDNA is destroyed through mitophagy processes; (b) mtDNA packaging into homoplasmic segregating units; and (c) focal replication, where specific populations of mtDNA are selected and replicated
[57][58][59][57,58,59]. A bottleneck in oocytes, which divide rapidly after being fertilized, generates a heterogeneous distribution of mtDNA subpopulations in daughter cells
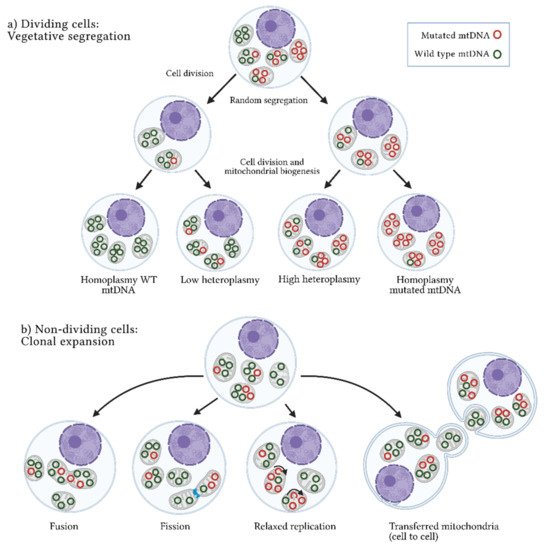
[49][58][60][61][49,58,60,61]. Two main mechanisms by which heteroplasmy differs among cells have been described: vegetative segregation and clonal expansion. In the first case, changes in allelic frequencies occur through the mitochondria random distribution during mitosis; in the second case, present in post-mitotic cells or non-dividing cells, heteroplasmy depends on the mtDNA replication rate and other cellular processes such as mitochondrial fusion and fission, as well as mtDNA transference between cells (
Figure 1)
[49][57][49,57]. Thus, heteroplasmy is an extraordinarily complex and dynamic event that results from various stochastic and deterministic cellular processes
[18][62][63][64][65][66][18,62,63,64,65,66]. Human pedigree analysis has shown that random genetic drift is involved in heteroplasmy and in combination with mitochondrial segregation, and the horizontal transfer of cell–cell mitochondria, modifies the allele frequencies of mtDNA over time. These frequencies are prone to increase or decrease depending on the effect of the variant
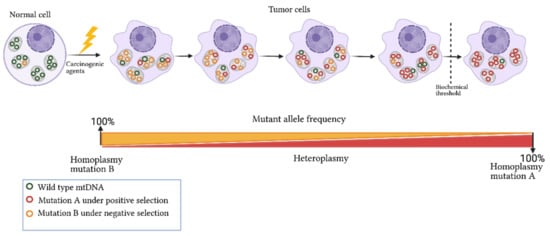
[67]. In this context, heteroplasmy is a transitional event between the disappearance or fixation of alleles that alters the mitochondrial function or confers advantageous biochemical conditions that promote cell division (
Figure 2)
[57][68][69][57,68,69]. A scan of heteroplasmic sites in different tissues showed the allele-specificity and tissue-specificity of some variants, showing evidence of the presence of positive and negative selection that promote or inhibit particular conditions of mitochondrial function
[70][71][70,71]. Transversions (point mutations that change a purine A or G by a pyrimidine T or C) and non-synonymous amino-acid changes were more commonly identified in a low heteroplasmic state, which reinforces the fact that, due to their potential pathogenic nature, these changes are selected negatively to reduce the damage of protein subunits involved in mitochondrial function. In contrast, a positive selection of somatic heteroplasmic sites was observed both in liver tissue and in specific genes such as NADH: ubiquinone oxidoreductase core subunit 5 (
MT-ND5), suggesting that these mutations provide specific biochemical features to the tissue
[70].
Figure 1. Heteroplasmy origin. Two mechanisms have been suggested to explain the origin and maintenance of the coexistence of wild-type and mutated mtDNA or heteroplasmy: (
a) Vegetative segregation. In dividing cells, mitochondria are randomly distributed among daughter cells, resulting in different proportions of mutated mtDNA molecules between them. (
b) Clonal expansion. In non-dividing cells, heteroplasmy is maintained through dynamic mitochondrial processes, such as fusion (union) or fission (separation) of mitochondria, relaxed replication (mtDNA replication independent of cell division), and intercellular mitochondrial transference. WT: wild-type; mtDNA: mitochondrial DNA molecules. Figure artwork created with
BioRender.com.
Figure 2. Dynamics of mitochondrial mutations in cancer. Exposure of a normal cell to carcinogenic agents leads to somatic mutation acquisition in mtDNA and changes in the germline mutation heteroplasmy. The frequency of acquired mutation can increase or decrease in tumor cells based on its nature and biological effect. For instance, if a mutation confers advantages to cell proliferation and survival, it will be selected and enriched in the tumor cell (mutation A). In contrast, a mutation compromising cell viability will be selected negatively (mutation B). Biochemical and phenotypic manifestation of the mtDNA mutations occurs only when a threshold level is exceeded. Figure artwork created with
BioRender.com.
Heteroplasmy is a common phenomenon, and low levels of heteroplasmy are commonly found in healthy tissues; the presence of potentially pathogenic mutations is not always translated into changes in the cell function or in a specific phenotype
[72][73][74][72,73,74]. The maintenance of normal mitochondrial function is explained by the existence of a “biochemical threshold”, a state where normal mtDNA buffers the damaging effect of mutated mtDNA molecules through the modulation of mitochondrial DNA replication, mitochondrial fission, and fusion processes
[73][75][73,75]. The threshold is tissue-specific, and the level of the mutated allele (heteroplasmy level) required to produce a pathological phenotype is dependent on the nature of the mutation
[57][74][76][57,74,76]. Many studies have reported that heteroplasmy levels of mutations positively correlate with ROS production and cellular damage that is potentially expressed as a wide range of clinical manifestations
[57][62][77][57,62,77]. For example, individuals carrying the A3243G mutation in heteroplasmy levels of 20%–30% commonly have type II diabetes mellitus; ranges from 50% to 80% of the mutated allele are manifested in myopathies, lactic acidosis, or cardiomyopathies; in a homoplasmic state, A3243G carriers develop Leigh syndrome
[78][79][78,79]. Additionally, the degree of heteroplasmy has been associated with different changes in the metabolic and epigenetic context
[78].