Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Florian Renosi and Version 2 by Dean Liu.
Differential diagnosis between Blastic pDC Neoplasm (BPDCN) and Acute Myeloid Leukemia with pDC expansion (pDC-AML) is particularly challenging, and genomic features can help in diagnosis. The genetic landscape of BPDCN is now well-defined, with important updates concerning MYC/MYC rearrangements, but also epigenetic defects and novel concepts in oncogenic and immune pathways.
- mature plasmacytoid dendritic cells proliferation
- acute myeloid leukemia
- RUNX1 mutation
1. Genetics of Blastic Plasmacytoid Dendritic Cell Neoplasms
Cytogenetic abnormalities are detected in 57 to 75% of BPDCN patients. Most of the karyotypes contain a wide spectrum of cytogenetic abnormalities, leading to a complex karyotype (CK) (≥3 aberrations) in more than 50% of cases [1][2][8,9]. Abnormal karyotypes of BPDCN show a high number of aberrations (mean = 6.8 per case). Using conventional cytogenetic and Fluorescent In Situ Hybridization (FISH)/multi-FISH approaches, a special and distinct cytogenetic signature of BPDCN have been described, showing various but recurrent chromosomal losses or deletions over gains. These abnormalities include 6 major recurrent chromosomal losses detected at high frequency among abnormal karyotype: 5q deletion (72%), 12p deletion (64%), 13q deletion or monosomy 13 (64%), 6q deletion (50%), 15q deletion or monosomy 15 (43%), and monosomy 9 (28%) [1][8]. Remarkably, among CK, careful examination revealed that three or more of these six chromosomal targets were associated in 50% of cases, defining a special cytogenetic signature for BPDCN (Table 1). These results have been confirmed by two independent studies [3][4][10,11].
These recurrent deletions were confirmed by chromosomal microarrays analyses, with losses of 9p21.3 (CDKN2A/CDKN2B), 12p13.2-p13.1 (CDKN1B, ETV6), 13q11-q21 (LATS2, RB1), 5q31 (NR3C1), or 7p12.2 (IKZF1) [1][2][5][8,9,12]. Despite this original pattern of recurrent abnormalities, there is no unique key genetic event in BPDCN. Indeed, at least four genes are particularly deleted and/or mutated: IKZF1, required for BPDCN differentiation [6][13]; RB1, potentially associated with transformation in the case of biallelic inactivation [7][14], ETV6, whose deletion would correspond to an early pathogenic event [4][11], and NR3C1, involved in the glucocorticoid metabolism [3][10]. The transcriptional activators MYC and MYB are also originally rearranged at high frequency in this neoplasm. Remarkably, the significant association of these alterations (i.e., loss of CDKN2A-B/9p21, CDKN1B/12p13, or RB1/13q14, rearrangement of MYC/8q24 or MYB/6q23) constitute the special and unique pattern frequently detected in BPDCN.
2. Genetics of Blastic Plasmacytoid Dendritic Cell Neoplasms
2.1. Deletions Involving Immune Genes
Interestingly, these recurrent deletions also involve genes related to the original function of pDC, i.e., immune response, especially losses of 6q23 (IFNGR1, TNFAIP3), 9p21.3 (cluster of IFNA genes), and 12p13.2-p13.1 (CLEC2B, CLEC4C, CLEC4E, TNFRSF1A) [8][15]. Thus, those deletions compromise the normal function of the cells of origin of BPDCN.2.2. Inactivation of Genes Encoding Cell-Cycle Inhibitors and Tumor Suppressor Genes
Similarly to other hematological malignancies, deletions inactivating Tumor Suppressor Genes (TSG), such as TP53 [9][10][11][12][16,17,18,19], responsible for genetic instability, are also found in BPDCN. The tumor suppressor gene ATM, mutated in lymphoproliferative syndromes [13][20], may also be mutated in BPDCN [12][19], while RB1, involved in the regulation of the G1/S cell cycle transition, is also frequently deleted [5][10][12][14][12,17,19,21]. Initially described in retinoblastoma, in which it modeled the principle of TSG [15][22], RB1 is also reported in chronic lymphocytic leukemia and Acute Lymphoblastic Leukemia (ALL) [16][23]. Additionally, the CDKN1B/CDKN2B/CDKN2A genes have a role in the G1/S transition, and their deletions are reported in some studies [2][5][10][12][17][9,12,17,19,24], similar to ALL [16][23] and lymphoma [18][25]. This alteration in cell cycle regulation could have a crucial role in the oncogenesis of BPDCN [10][17].2.3. Recurrent Deletions in 5q31
Deletion in 5q are particularly recurrent in BPDCN, constituting a specific defect compared to other hematological malignancies [3][19][10,26]. In the 5q23.3 Common Deleted Region (CDR), HINT1 was first proposed to be a key gene [19][26]. Indeed, HINT1 encodes a homodimeric purine phosphoramidase, suggesting a transcriptional modulatory role. Moreover, HINT1 deficiency would impair ATM function and thus DNA repair [19][26]. On the other hand, the 5q31 locus would also be a key region on chromosome 5, with deletions delineating a group of unfavorable prognostic impacts [3][10]. The glucocorticoid receptor gene NR3C1 was found to be recurrently deleted, leading to haploinsufficiency and decreased glucocorticoid receptor activity [3][10]. The deletions impact the polycomb complex, in particular EZH2, with dysregulation of the HOXA locus and plasmacytoid dendritic differentiation.2.4. Deletions of Transcription Factors
Similarly to other hematological neoplasms, transcription factor are particularly impacted in BPDCN. ETV6 (TEL) is frequently mutated or deleted [1][5][8][9][10][17][8,12,15,16,17,24], contrasting with classical defects in other leukemia where translocations are more frequent, including t(12;21)(p13;q22) ETV6::RUNX1 in B-cell Acute Lymphoblastic Leukemia (B-ALL) [20][27], t(5;12)(q32;p13) ETV6::PDGFRB, t(9;12)(p24;p13) ETV6::JAK2 or t(4;12) (q12;p13) ETV6::PDGFRA in hypereosinophilic syndromes, and other translocations in rare cases of Acute Myeloid Leukemia (AML) [21][28]. ETV6 invalidations are, however, known in leukemia: somatic mutations of ETV6 remain rare in AML [21][28], but germline mutations are also possible in the context of thrombocytopenia predisposing to AML, Myelodysplastic Syndromes (MDS), Chronic MyeloMonocytic Leukemia (CMML), B-ALL, or multiple myeloma [22][1]. The IKAROS family (IKZF1/2/3) is also frequently deleted [2][9][14][23][9,16,21,29], similarly to ALL [24][30], where it compromises lymphoid differentiation [25][31]. ZEB2 may also be altered in BPDCN [26][32]. This transcription factor is involved in the commitment and lineage fidelity of myeloid and lymphoid cells at various stages of hematopoiesis and is thought to play a key role in the development of various types of AML, ALL, and lymphoma [27][33]. While translocations involving transcription factors have been widely described in ALL and AML, recurrent rearrangements in BPDCN were rare before 2017. Indeed, KMT2A (MLL) rearrangements had previously been described in rare cases of CD4+ CD56+ neoplams identified as BPDCN (KMT2A::ENL and KMT2A::MLLT1) [28][29][34,35], but these descriptions in BDPCN have been challenged because these reported cases do not fulfill the current diagnostic criteria of BPDCN. Indeed, these cases could correspond to CD4+ CD56+ AML, because they constitute a delicate differential diagnosis for BPDCN [30][36].2.5. Recurrent MYC Rearrangements
Initially, the translocation t(6;8)(p21;q24) was sporadically reported by several studies, using conventional karyotyping. Since 2018, MYC rearrangements (8q24) have been largely described in approximately 30% of BPDCN [23][31][32][33][29,37,38,39], with a more frequent immunoblastoid morphology and sometimes a CD56-negative phenotype [31][34][37,40]. These MYC abnormalities bring BPDCN closer to high-grade B lymphoma [2][6][31][35][9,13,37,41], but the gene partners are really different, with specific partners that could point towards pDC differentiation. Indeed, among MYC rearranged cases of BPDCN, Sakamoto et al. confirmed the high prevalence of the t(6;8)—detected in 56% of cases involving the RUNX2 locus at 6p21. Interestingly, Kubota et al. showed that the t(6;8) juxtaposes the promoter of MYC to the pDCs-specific RUNX2 super-enhancer, leading to overexpression of MYC. In this recurrent t(6;8)(p21;q24), both MYC and RUNX2 are dysregulated, and cooperate together to promote survival and proliferation of the BPDCN cells. Remarkably, RUNX2 is physiologically involved in differentiation and migration of pDCs and plays a dominant role in controlling transcription networks in BPDCN [36][42]. Other partners of MYC have been sporadically reported but not clearly identified (i.e., 2p12, Xq24, 3p25, 14q32). It remains to be determined if MYC rearrangement could constitute a primary or secondary genetic event in BPDCN. In this way, the t(6;8)(p21;q24) cannot be considered as a specific genetic abnormality of BPDCN because it has been reported in follicular lymphoma [37][43]. Lastly, a unique study showed the adverse impact of MYC rearrangement, and this prognostic impact still needs to be confirmed by further independent studies [31][37].2.6. Recurrent MYB Rearrangements
In 2017, other recurrent rearrangements were described in nine of fourteen patients, including five children [14][21]. Remarkably, all five children included in this series had a MYB rearrangement. Of note, the previous largest report of pediatric BPDCN cases exhibited several cases with 1q and/or 6q abnormalities, or translocation t(1;6)(q21;q23) [38][44]. These observations reveal a striking link between pediatric BPDCN and MYB rearrangement. MYB rearrangements create fusion transcripts between MYB and various partner genes (ZFAT/8q24, PLEKHO1/1q21, DCPS/11q24, miR-3134/3p25) [14][21]. The chimeric transcripts retain the MYB transactivation domain and disrupt its negative regulatory domain, which allows the maintenance of the MYB transcriptional activity. Indeed, functional analysis of MYB fusions revealed the activation of MYB target genes as a result of induced MYB activation [14][21]. MYB is a nuclear-localized transcriptional activator in hematopoietic cells that interacts with the C/EBP complex to stimulate the transcriptional activity of MYC, BCL2, c-KIT, c-ERBB2, and other targets (Figure 1). Its expression progressively decreases during cell differentiation, with high activity in hematopoietic stem cells and activated T-cells.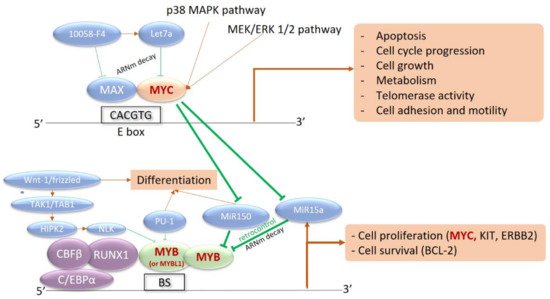
Figure 1. Interaction network between MYB, MYC, and their transcriptional targets. MYC is a strong transcriptional activator, dependent on the intracellular phosphorylation cascade signaling pathways of Mitogen-Activated Protein Kinases (MAPK) and Extracellular signal-Regulated Kinases (ERK) (p38 MAPK and MEK/ERK pathways 1 and 2). Activation of MYC induces the transcription of numerous target genes involved in proliferation, division, metabolism, and cell motility, as well as apoptosis. MYC also inhibits microRNAs (MiR150 and MiR15a) that are capable of silencing MYB expression in the basal state. Activation of MYC therefore induces activation of MYB, involved in the core binding factor (CBF) complex with CBFB, RUNX1, and CEBPA. MYB is also a transcriptional activator recognizing multiple nucleotide sequences, in a complex with CEBP. The targeted genes are involved in survival with BCL-2 and cell proliferation with c-KIT, c-ERBB2, and especially MYC. This results in an activation loop between the two transcriptional activators: BS, MYB Binding Site with MYB Recognition Element.
2.7. Mutation Landscape
2.7.1. A Myeloid-like Profile
In addition to these cytogenetic defects, the mutation landscape of BPDCN has been particularly studied. However, given the rarity of BPDCN, only small cohorts have been studied so far (less than 30 cases), and usually by targeted high-throughput sequencing. Nevertheless, 22 Whole Exome Sequencing (WES) have been performed [7][9][44][14,16,50]. On a first stratum, the mutation landscape is quite similar to myeloid neoplasms [45][46][47][48][49][50][51,52,53,54,55,56], with a high prevalence of mutations involving epigenetics (TET2, ASXL1) and splicing (ZRSR2, SRSF2, U2AF1) [9][11][51][52][53][16,18,57,58,59]. These mutations rather suggest an early process before leukemic transformation, as is well described in AML, disturbing the DNA methylation balance, modifying chromatin access and the splicing processes [54][60]. In multistage leukemogenesis models, epigenetics and splicing mutations would be present from the pre-leukemic stages [55][61], and their frequency increases with age. Of note, mutations of TET2 are found in 40 to 60% of cases [9][11][53][16,18,59]. Interestingly, loss-of-function of ZRSR2 impairs pDC activation and apoptosis after inflammatory stimuli with intron retention, promoting pDC expansion. Of note, being located on the X chromosome, this enrichment of ZRSR2 in BPDCN fits well with its predominance in males [52][58]. Although mutations of NPM1 were initially described in BPDCN [9][16], this has not been confirmed since, and this is not consistent with the nature of these mutations defining a mutually exclusive subtype of AML [22][1]. In contrast, sub-clonal mutations of signaling pathways can be found in 5 to 20% of BPDCN, especially FLT3, KIT, KRAS, and NRAS mutations [9][12][14][26][44][45][53][56][57][58][16,19,21,32,50,51,59,62,63,64]. This profile is close to that of CMML and is consistent with a common clonal origin of BPDCN and CMML cells demonstrated in a few patients suffering from the two neoplasms [7][45][14,51]. The leukemic model would include shared epigenetic mutations, with secondary emergence of a BPDCN clone and another clone leading to CMML [48][59][54,65] or AML [60][61][62][63][66,67,68,69].2.7.2. Some Lymphoid-like Features
Associated with these “myeloid-like”, key deleted transcription factors or tumor suppressor genes IKZF1, ETV6, RB1, ATM, and TP53 can also be mutated in some cases (5–10%), also resulting in an invalidation [2][4][6][7][11][64][65][9,11,13,14,18,70,71]. Notably, biallelic invalidations of ETV6 argue for a primordial early event, possibly overexpressing the BPDCN oncogene TCL1A [4][8][66][11,15,72]. IKZF1 loss-of-function, either by deletion or mutation, would lead to the increased cell interactions in BPDCN. BPDCN also exhibit KMT2D and SYNE1 mutations or losses, previously reported in follicular lymphoma [6][64][13,70]. Overall, the most characteristic feature of BPDCN would be that combination of myeloid-like and lymphoid-like abnormalities (Figure 2).
Figure 2. Genomics and transcriptional landscape of BPDCN. The genomic landscape of BPDCN include a combination of myeloid-like and lymphoid-like mutations and cytogenetic defects within a complex landscape, with frequent complex karyotypes. The transcriptional program of BPDCN is made of a diversity of original factors: RUNX2, MYB, IFN pathway, neural processes, cholesterol metabolism, corticoresistance factor, and original oncogenic factors.