Coronaviruses represent a diverse family of enveloped positive-sense single stranded RNA viruses. COVID-19, caused by Severe Acute Respiratory Syndrome Coronavirus-2, is a highly contagious respiratory disease transmissible mainly via close contact and respiratory droplets which can result in severe, life-threatening respiratory pathologies. It is understood that glutathione, a naturally occurring antioxidant known for its role in immune response and cellular detoxification, is the target of various proinflammatory cytokines and transcription factors resulting in the infection, replication, and production of reactive oxygen species. This leads to more severe symptoms of COVID-19 and increased susceptibility to other illnesses such as tuberculosis.
- glutathione
- coronavirus
- SARS-CoV-2
- COVID-19
1. SARS-CoV-2
2. Evolving into Multiple Variants
3. Increased Susceptibility to COVID-19 among Risk Groups
4. Pathogenesis of the Disease Induced by Cytokine Storm, Increased IL-6, Decreased Glutathione
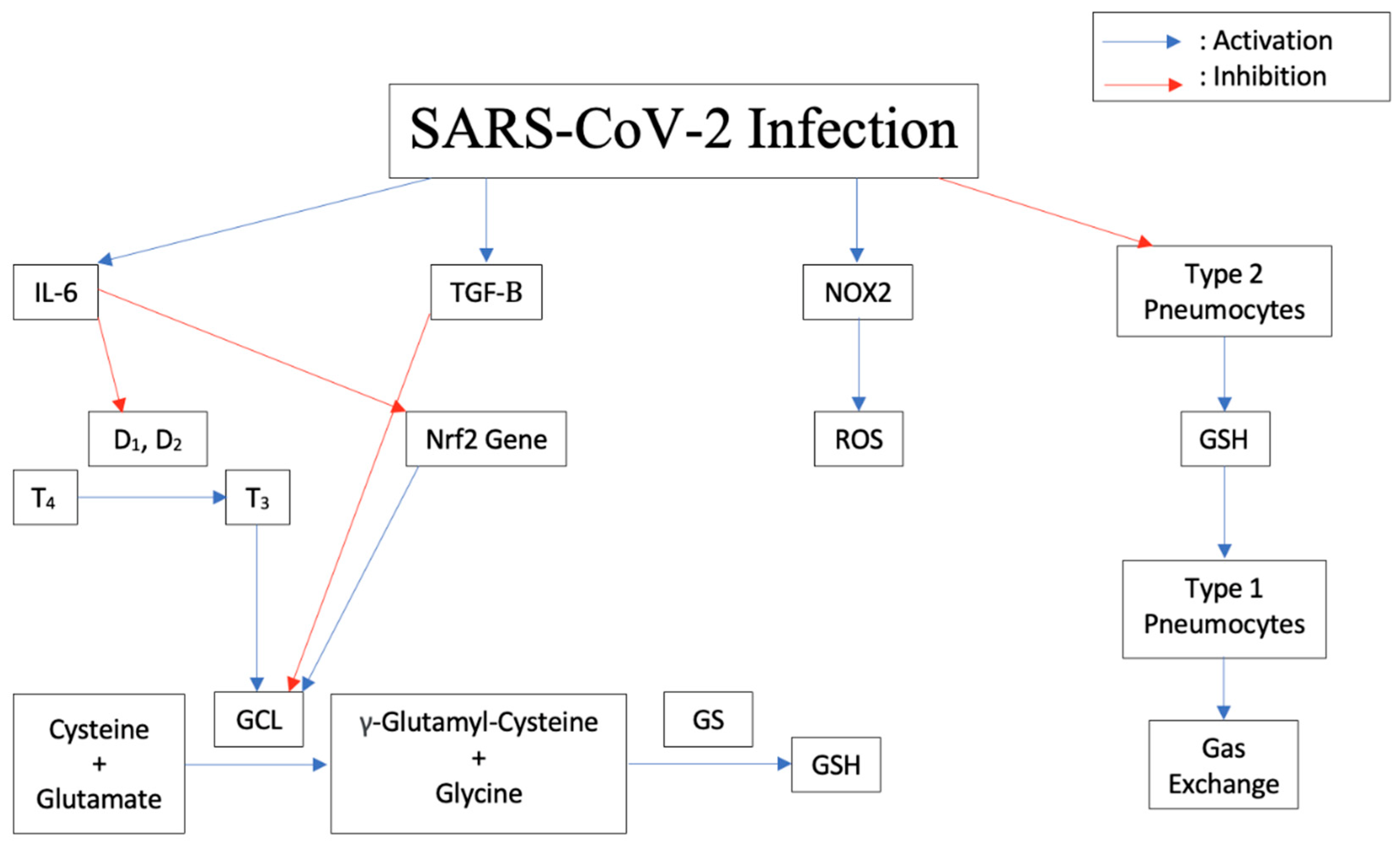
5. Increased Susceptibility to TB Due to Decrease in Glutathione
6. Modulation of Expression of Antioxidant Genes
References
- Ochani, R.; Asad, A.; Yasmin, F.; Shaikh, S.; Khalid, H.; Batra, S.; Sohail, M.R.; Mahmood, S.F.; Ochani, R.; Arshad, M.H.; et al. COVID-19 pandemic: From origins to outcomes. A comprehensive review of viral pathogenesis, clinical manifestations, diagnostic evaluation, and management. Infez. Med. 2021, 29, 20–36. Available online: https://pubmed.ncbi.nlm.nih.gov/33664170/ (accessed on 14 April 2022).
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733.
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170.
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2020, 3, 237–261.
- Pascarella, S.; Ciccozzi, M.; Bianchi, M.; Benvenuto, D.; Cauda, R.; Cassone, A. The electrostatic potential of the Omicron variant spike is higher than in Delta and Delta-plus variants: A hint to higher transmissibility? J. Med. Virol. 2021, 94, 1277–1280.
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166.
- World Health Organization. Tracking SARS-CoV-2 Variants. 2022. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 10 June 2022).
- Kemp, S.; Collier, D.; Datir, R.; Ferreira, I.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. Neutralizing antibodies in Spike mediated SARS-CoV-2 adaptation. medRxiv 2020.
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020.
- Centers for Disease Control and Prevention. Emerging SARS-CoV-2 Variants. COVID-19. 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html (accessed on 10 June 2022).
- Tellez, D.; Dayal, S.; Phan, P.; Mawley, A.; Shah, K.; Consunji, G.; Tellez, C.; Ruiz, K.; Sabnis, R.; Dayal, S.; et al. Analysis of COVID-19 on Diagnosis, Vaccine, Treatment, and Pathogenesis with Clinical Scenarios. Clin. Pract. 2021, 11, 309–321.
- Ejaz, H.; Alsrhani, A.; Zafar, A.; Javed, H.; Junaid, K.; Abdalla, A.E.; Abosalif, K.O.A.; Ahmed, Z.; Younas, S. COVID-19 and comorbidities: Deleterious impact on infected patients. J. Infect. Public Health 2020, 13, 1833–1839.
- Abu-Farha, M.; Al-Mulla, F.; Thanaraj, T.A.; Kavalakatt, S.; Ali, H.; Abdul Ghani, M.; Abubaker, J. Impact of Diabetes in Patients Diagnosed with COVID-19. Front. Immunol. 2020, 11, 576818.
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-Modulating Agents in the Treatment of Viral Infections. Int. J. Mol. Sci. 2020, 21, 4084.
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003, 17, 758–760.
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell. Microbiol. 2014, 17, 131–145.
- Celestino, I.; Checconi, P.; Amatore, D.; De Angelis, M.; Coluccio, P.; Dattilo, R.; Alunni Fegatelli, D.; Clemente, A.M.; Matarrese, P.; Torcia, M.G.; et al. Differential Redox State Contributes to Sex Disparities in the Response to Influenza Virus Infection in Male and Female Mice. Front. Immunol. 2018, 9, 1747.
- Wajner, S.M.; Goemann, I.M.; Bueno, A.L.; Larsen, P.R.; Maia, A.L. IL-6 promotes nonthyroidal illness syndrome by blocking thyroxine activation while promoting thyroid hormone inactivation in human cells. J. Clin. Investig. 2011, 121, 1834–1845.
- Dasgupta, A.; Das, S.; Sarkar, P.K. Thyroid hormone promotes glutathione synthesis in astrocytes by up regulation of glutamate cysteine ligase through differential stimulation of its catalytic and modulator subunit mRNAs. Free Radic. Biol. Med. 2007, 42, 617–626.
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.C.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.R.S.; et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69.
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.S.; Drummond, G.R.; Selemidis, S. Inhibition of Nox2 Oxidase Activity Ameliorates Influenza A Virus-Induced Lung Inflammation. PLoS Pathog. 2011, 7, e1001271.
- Forman, H.J. Glutathione—From antioxidant to post-translational modifier. Arch. Biochem. Biophys. 2016, 595, 64–67.
- Diotallevi, M.; Checconi, P.; Palamara, A.T.; Celestino, I.; Coppo, L.; Holmgren, A.; Abbas, K.; Peyrot, F.; Mengozzi, M.; Ghezzi, P. Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties. Front. Immunol. 2017, 8, 1239.
- Checconi, P.; Limongi, D.; Baldelli, S.; Ciriolo, M.R.; Nencioni, L.; Palamara, A.T. Role of Glutathionylation in Infection and Inflammation. Nutrients 2019, 11, 1952.
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox Signal. 2011, 15, 593–606.
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417.
- Forcina, L.; Miano, C.; Scicchitano, B.M.; Rizzuto, E.; Berardinelli, M.G.; De Benedetti, F.; Pelosi, L.; Musarò, A. Increased Circulating Levels of Interleukin-6 Affect the Redox Balance in Skeletal Muscle. Oxidative Med. Cell. Longev. 2019, 2019, 3018584.
- Ghazavi, A.; Ganji, A.; Keshavarzian, N.; Rabiemajd, S.; Mosayebi, G. Cytokine profile and disease severity in patients with COVID-19. Cytokine 2021, 137, 155323.
- He, L.; Ding, Y.; Zhang, Q.; Che, X.; He, Y.; Shen, H.; Wang, H.; Li, Z.; Zhao, L.; Geng, J.; et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 2006, 210, 288–297.
- Liu, R.-M.; Vayalil, P.K.; Ballinger, C.; Dickinson, D.A.; Huang, W.-T.; Wang, S.; Kavanagh, T.J.; Matthews, Q.L.; Postlethwait, E.M. Transforming growth factor β suppresses glutamate-cysteine ligase gene expression and induces oxidative stress in a lung fibrosis model. Free Radic. Biol. Med. 2012, 53, 554–563.
- Morris, D.; Guerra, C.; Donohue, C.; Oh, H.; Khurasany, M.; Venketaraman, V. Unveiling the Mechanisms for Decreased Glutathione in Individuals with HIV Infection. Clin. Dev. Immunol. 2012, 2012, 734125.
- Hu, Q.; Guan, H.; Sun, Z.; Huang, L.; Chen, C.; Ai, T.; Pan, Y.; Xia, L. Early CT features and temporal lung changes in COVID-19 pneumonia in Wuhan, China. Eur. J. Radiol. 2020, 128, 109017.
- Lechowicz, K.; Drożdżal, S.; Machaj, F.; Rosik, J.; Szostak, B.; Zegan-Barańska, M.; Biernawska, J.; Dabrowski, W.; Rotter, I.; Kotfis, K. COVID-19: The Potential Treatment of Pulmonary Fibrosis Associated with SARS-CoV-2 Infection. J. Clin. Med. 2020, 9, 1917.
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422.
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686.
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140.
- Agrati, C.; Sacchi, A.; Bordoni, V.; Cimini, E.; Notari, S.; Grassi, G.; Casetti, R.; Tartaglia, E.; Lalle, E.; D’Abramo, A.; et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ. 2020, 27, 3196–3207.
- van Klaveren, R.J.; Demedts, M.; Nemery, B. Cellular glutathione turnover in vitro, with emphasis on type II pneumocytes. Eur. Respir. J. 1997, 10, 1392–1400.
- Rahman, I.; MacNee, W. Oxidative stress and regulation of glutathione in lung inflammation. Eur. Respir. J. 2000, 16, 534.
- Gibson, P.G.; Qin, L.; Puah, S.H. COVID-19 acute respiratory distress syndrome (ARDS): Clinical features and differences from typical pre-COVID-19 ARDS. Med. J. Aust. 2020, 213, 54–56.e1.
- Tobin, M.J.; Laghi, F.; Jubran, A. Why COVID-19 Silent Hypoxemia Is Baffling to Physicians. Am. J. Respir. Crit. Care Med. 2020, 202, 356–360.
- Singh, M.; Vaughn, C.; Sasaninia, K.; Yeh, C.; Mehta, D.; Khieran, I.; Venketaraman, V. Understanding the Relationship between Glutathione, TGF-β, and Vitamin D in Combating Mycobacterium tuberculosis Infections. J. Clin. Med. 2020, 9, 2757.
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxidative Med. Cell. Longev. 2016, 2016, 8910396.
- Shatrov, V.A.; Ratter, F.; Gruber, A.; Dröoge, W.; Lehmann, V. HIV Type 1 Glycoprotein 120 Amplifies Tumor Necrosis Factor-Induced NF-kB Activation in Jurkat Cells. AIDS Res. Hum. Retrovir. 1996, 12, 1209–1216.
- Gu, Y.; Wu, R.F.; Xu, Y.C.; Flores, S.C.; Terada, L.S. HIV Tat Activates c-Jun Amino-terminal Kinase through an Oxidant-Dependent Mechanism. Virology 2001, 286, 62–71.
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 2013, 4, e850.
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15.
- Ly, J.; Lagman, M.; Saing, T.; Singh, M.K.; Tudela, E.V.; Morris, D.; Anderson, J.; Daliva, J.; Ochoa, C.; Patel, N.; et al. Liposomal Glutathione Supplementation Restores TH1 Cytokine Response to Mycobacterium tuberculosis Infection in HIV-Infected Individuals. J. Interferon Cytokine Res. 2015, 35, 875–887.
- Valdivia, A.; Ly, J.; Gonzalez, L.; Hussain, P.; Saing, T.; Islamoglu, H.; Pearce, D.; Ochoa, C.; Venketaraman, V. Restoring Cytokine Balance in HIV-Positive Individuals with Low CD4 T Cell Counts. AIDS Res. Hum. Retrovir. 2017, 33, 905–918.
- Chumburidze-Areshidze, N.; Kezeli, T.; Avaliani, Z.; Mirziashvili, M.; Avaliani, T.; Gongadze, N. The Relationship between Type-2 Diabetes and Tuberculosis. Georgian Med. News 2020, 300, 69–74.
- Calabrese, V.; Cornelius, C.; Leso, V.; Trovato-Salinaro, A.; Ventimiglia, B.; Cavallaro, M.; Scuto, M.; Rizza, S.; Zanoli, L.; Neri, S.; et al. Oxidative stress, glutathione status, sirtuin and cellular stress response in type 2 diabetes. Biochim. Biophys. Acta—Mol. Basis Dis. 2012, 1822, 729–736.
- Lee, C. Therapeutic Modulation of Virus-Induced Oxidative Stress via the Nrf2-Dependent Antioxidative Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 6208067.
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930.
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The Antioxidant Defense System Keap1-Nrf2 Comprises a Multiple Sensing Mechanism for Responding to a Wide Range of Chemical Compounds. Mol. Cell. Biol. 2009, 29, 493–502.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86.
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase to Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139.
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and Electrophilic Stresses Activate Nrf2 through Inhibition of Ubiquitination Activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229.
- Solis, W.A.; Dalton, T.P.; Dieter, M.Z.; Freshwater, S.; Harrer, J.M.; He, L.; Shertzer, H.G.; Nebert, D.W. Glutamate–cysteine ligase modifier subunit: Mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 2002, 63, 1739–1754.
- Cho, H.Y.; Reddy, S.P.; DeBiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343.
- Das, B.N.; Kim, Y.W.; Keum, Y.S. Mechanisms of Nrf2/Keap1-Dependent Phase II Cytoprotective and Detoxifying Gene Expression and Potential Cellular Targets of Chemopreventive Isothiocyanates. Oxidative Med. Cell. Longev. 2013, 2013, 839409.
- Jin, C.H.; So, Y.K.; Han, S.N.; Kim, J.B. Isoegomaketone Upregulates Heme Oxygenase-1 in RAW264.7 Cells via ROS/p38 MAPK/Nrf2 Pathway. Biomol. Ther. 2016, 24, 510–516.
- Herzenberg, L.A.; De Rosa, S.C.; Dubs, J.G.; Roederer, M.; Anderson, M.T.; Ela, S.W.; Deresinski, S.C.; Herzenberg, L.A. Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA 1997, 94, 1967–1972.
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual Role of NOX2 in Respiratory Syncytial Virus- and Sendai Virus-Induced Activation of NF-κB in Airway Epithelial Cells. J. Immunol. 2008, 180, 6911–6922.
- Komaravelli, N.; Ansar, M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces NRF2 degradation through a promyelocytic leukemia protein - ring finger protein 4 dependent pathway. Free Radic. Biol. Med. 2017, 113, 494–504.
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory Syncytial Virus Induces Oxidative Stress by Modulating Antioxidant Enzymes. Am. J. Respir. Cell Mol. Biol. 2009, 41, 348–357.
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants 2020, 9, 624.
- Zhang, J.; Cruz-cosme, R.; Zhuang, M.W.; Liu, D.; Liu, Y.; Teng, S.; Wang, P.H.; Tang, Q. Correction: A systemic and molecular study of subcellular localization of SARS-CoV-2 proteins. Signal Transduct. Target. Ther. 2021, 6, 192.
- De Michele, M.; d’Amati, G.; Leopizzi, M.; Iacobucci, M.; Berto, I.; Lorenzano, S.; Mazzuti, L.; Turriziani, O.; Schiavo, O.G.; Toni, D. Evidence of SARS-CoV-2 spike protein on retrieved thrombi from COVID-19 patients. J. Hematol. Oncol. 2022, 16, 108.
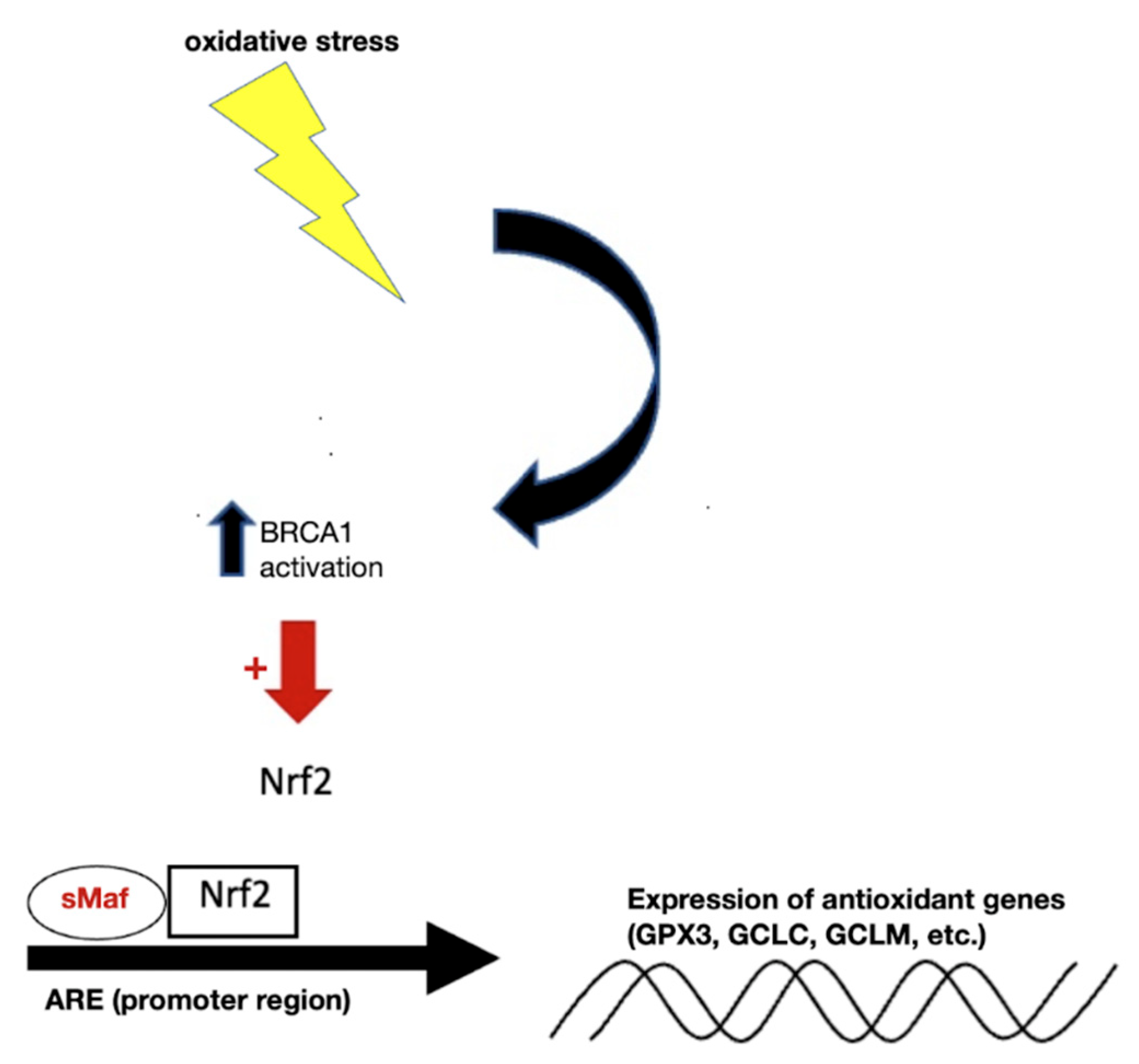
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 Induces Antioxidant Gene Expression and Resistance to Oxidative Stress. Cancer Res. 2004, 64, 7893–7909.
- Kovacic, P.; Jacintho, J. Mechanisms of Carcinogenesis Focus on Oxidative Stress and Electron Transfer. Curr. Med. Chem. 2001, 8, 773–796.
- Lorente, L.; Martín, M.M.; González-Rivero, A.F.; Pérez-Cejas, A.; Cáceres, J.J.; Perez, A.; Ramos-Gómez, L.; Solé-Violán, J.; Ramos, J.A.M.Y.; Ojeda, N.; et al. DNA and RNA Oxidative Damage and Mortality of Patients with COVID-19. Am. J. Med. Sci. 2021, 361, 585–590.
- Grifoni, E.; Valoriani, A.; Cei, F.; Lamanna, R.; Gelli, A.M.G.; Ciambotti, B.; Vannucchi, V.; Moroni, F.; Pelagatti, L.; Tarquini, R.; et al. Interleukin-6 as prognosticator in patients with COVID-19. J. Infect. 2020, 81, 452–482.