1. Aminopyrazoles
TWe start this chapter
is strarted by discussing synthetical procedures used to obtain 5-aminopyrazoles, whose chemistry has been well documented for a long time
[1][2][4,22].
The examples are supposed to be exposedWe wanted to expose examples from the last five years; however, other earlier key examples are worth mentioning. For instance,
t we have decided to mention the work reported in 2016 by Kallman et al.
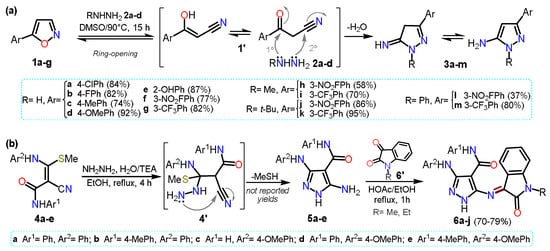
[3] will be mentioned[25], which is not a standard procedure for accessing 5-aminopyrazoles. Specifically, the authors reported a regioselective synthesis of aminopyrazoles from isoxazoles
1a–g as they are synthetic equivalents of ketonitriles
1′. The reaction proceeds via ring-opening, generating a ketonitrile
1′ intermediate that then reacts with hydrazine derivatives
2a–d to form the respective cyclocondensation product
3a–m (
Scheme 1Scheme 2a).
Scheme 12.
Synthesis of 5-aminopyrazoles by (
a
) isoxazole ring-opening and (
b
) from enaminonitriles.
In 2021, Hassan and co-workers
[4][26] reported the synthesis of pyrazole-oxindole hybrid systems
6a–g by the condensation reaction of 5-aminopyrazoles
5a–e with
N-substituted isatin
6′ (
Scheme 1Scheme 2b). Heteroamines
5a–e were obtained by the cyclocondensation reaction of
N-aryl-3-(arylamino)-2-cyano-3-(methylthio)acrylamides
4a–e with hydrazine hydrate (
2a). Intermediates
5 are substituted with arylamines and amides at positions 3 and 4, making it possible for the core to have a wide range of post-functionalizations.
HIn this case
rein, final products
6a–j were used for in vitro cytotoxicity assays against four human cancer-type cells; it is important to note that in the examples about 5-aminopyrazoles synthesis mentioned above, and in most others involving ketonitriles or enaminonitriles as 1,3-bis-electrophilic substrates, it is necessary to have an easy displacement group on the Cβ of the substrate to generate the required unsaturation in the product.
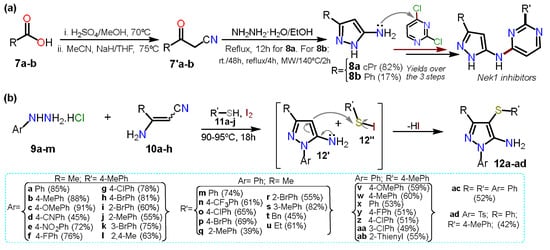
On the other hand, Pilakowski et al.
[5][27] synthesized 5-alkyl-3-amino-1
H-pyrazoles
8a–b starting from carboxylic acids
7a–b (
Scheme 2Scheme 3a). First, an esterification reaction was developed, and the respective ester was treated with sodium hydride and acetonitrile to form the corresponding ketonitrile
7′. Next, this intermediate was treated with hydrazine hydrate to obtain the desired products
8a–b, which were then coupled to dichloropyrimidine to yield N-substituted pyrazoles tested as Nek1-inhibitors. The low yield of
8b (17%) versus
8a (82%) is due to the last step, where the authors subjected the reaction with substrate
7′b to different conditions; they possibly wanted to obtain the product as the expected light yellow solid; however, they only managed to isolate it as a viscous orange oil.
Scheme 23.
5-Aminopyrazole synthesis forms (
a
) carboxylic acids and (
b
) enaminonitriles.
In another recent approach, Annes et al.
[6][28] reported a free-metal and free-solvent multicomponent synthesis mediated by iodine to obtain aminopyrazole-thioether derivatives
12a–ad in the range of 39–91% yield. The multicomponent reaction comprises substituted hydrazines
9a–m, nitriles
10a–h benzenethiols
11a–j. Reagents
9a–m and
10a–h undergo a Michael reaction in the presence of Lewis acid, followed by intramolecular cyclization with the elimination of ammonia to afford 5-aminopyrazoles
12′. At the same time, iodine reacts with
11 to form the electrophilic derivative
12″. Finally, the C-S bond was formed via an electrophilic aromatic substitution (EAS) reaction on
12′. The reaction scope was studied, including diverse aromatic and aryl groups at positions 1, 3, and 4 (
Scheme 2Scheme 3b); this reaction proceeded with a wide range of substrates; however, the best yields are obtained when the aminopyrazole
12′ has an electron releasing group (ERG) or the electrophilic reagent
12″ has an electron-withdrawing group (EWG).
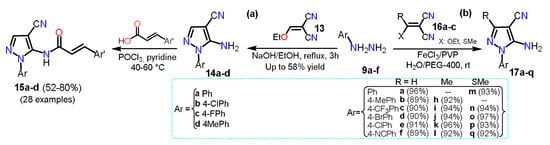
In 2018, Ren et al.
[7][29] synthesized 5-amino-1-arylpyrazole-4-carbonitriles
14a–d starting from a mixture of arylhydrazine hydrochloride
9a, 2-(ethoxymethyl)malononitrile
13, ethanol, and sodium hydroxide via a classical cyclocondensation reaction (
Scheme 3Scheme 4a). With 5-aminopyrazoles
14a–d in hand, the authors transformed them into the carboxamide derivatives
15a–d, which then were evaluated against three fungal strains and as inhibitory compounds against succinate dehydrogenase. A year later, Elnagdy and Sarma
[8][30] reported a homogenous catalytic system using FeCl
3/PVP and green solvent water/PEG-400 to synthesize 4-amino-1-aryl-1
H-pyrazole-4-carbonitriles
17a–q using a cyclocondensation reaction of arylhydrazines
9a–f with malononitrile derivatives
16a–c. A mixture of FeCl
3 and polyvinyl pyrrolidine (PVP) was used to accelerate the addition of
9a–f to the double bond of
16a–c; then, an intramolecular cyclization allows the formation of products
17a–q in up to 97% yield with reaction times of 2–4 h (
Scheme 3Scheme 4b).
Scheme 34.
Pyrazole synthesis from malononitrile derivatives (
a
)
13
and (
b
)
16a–c
.
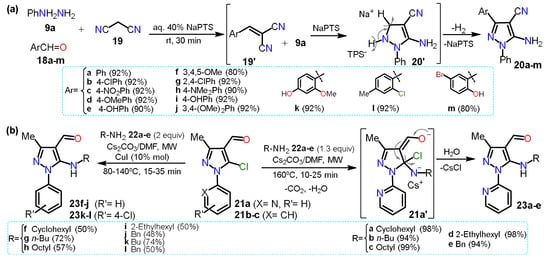
In 2020, Sapkal and Kamble
[9][31] obtained 5-aminopyrazole-4-carbonitriles
20a–m using a green protocol based on a three-component cyclocondensation of phenylhydrazine
9a, aldehyde derivatives
18a–m, and malononitrile (
19) adding sodium
p-toluenesulfonate (NaPTS) as a catalyst in aqueous media (
Scheme 5a). The authors mentioned that NaPTS was used as a hydrotrope that helps increase the solubility of poorly soluble organic compounds in water. First, water hydrates the hydrotrope head groups, decreasing their electrostatic attraction. Both head groups move apart, displacing water molecules interacting with hydrophobic parts; this action helps the reactant molecules interact, enhancing the reaction on aqueous media. The reaction mechanism starts with the nucleophilic attack of
19 on the electrophilic carbon of arylaldehydes
18a–m to form arlylidenemalononitrile derivatives
19′. Then,
9a proceeds by a nucleophilic attack over the double bond of
19′, and finally, the addition intermediate undergoes intramolecular cyclization to afford products
20a–m. Although the authors mention that the presence of NaPTS favors the reaction by increasing the solubility of reactants,
it iswe believe
d that it mainly helps in the product aromatization step (
20′ in
Scheme 5a) as
19′ do not possess a leaving group. For this synthesis, the substituent electronic effects do not influence the yields and scope of the reaction.
Scheme 45.
Synthesis of 5-aminopyrazoles from (
a
) malononitriles and (
b
) 5-chloropyrazoles.
This section started with a “non-classic” method to obtain 5-aminopyrazoles, and in 2015, another not classic strategy was described via a nucleophilic aromatic substitution (NAS) reaction on 5-chloropyrazole derivatives. Specifically, 5-(
N-alkyl)aminopyrazoles
23a–e were synthesized in high yields via the microwave-assisted reaction between 5-chloro-4-formylpyrazoles
21a–c and primary alkylamines
22a–e [10][32]. The reaction was possible because the amine nucleophilicity is favored under MW by the cesium effect and the substrate
21a has a 2-pyridyl group at position 1, which is a strong EWG; however, using the
N-aryl substituted substrate
21b–c, the reaction needs harsh conditions and even CuI as a catalyst to form
23f–l; this reaction type has been scarcely studied since the pyrazole ring exhibits a moderate π-excedent character, which disfavors the initial nucleophilic attack. Therefore, these results corroborate the difficulty of the NAS reaction on pyrazole derivatives justifying its limited study (
Scheme 4Scheme 5b).
2. Acylpyrazoles
2. Acylpyrazoles
2.1. Formylpyrazoles
Formylpyrazoles are strategic intermediates in obtaining a wide range of biologically active compounds, with the 4-formyl derivatives being more usual; they possess a high synthetical versatility allowing them a plethora of reactions for the insertion of more functional groups.
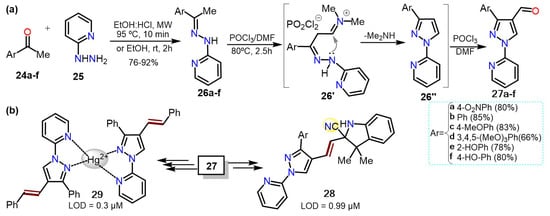
TheOur research group has reported the synthesis of 3-aryl-1-(pyridin-2-yl)-1
H-pyrazole-4-carbaldehydes
27a–f via Vilsmeier-Haack cyclization-formilation of different hydrazones
26a–f, which were generated from acetophenones
24a–f and 2-hydrazinylpyridine (
25). Precursor
26 was transformed in the 1,3-biselectrophilic intermediate
26′ under Vilsmeier-Haack conditions. Subsequently,
26′ is cyclocondensed to pyrazole
26″, which is finally formylated to deliver 4-formylpyrazole
27 in 66–85% yields (
Scheme 5Scheme 6a).
Scheme 56.
Synthesis of (
a
) 4-formyl-1-(2-pyridyl)pyrazoles and (
b
)chemosensors based on pyrazoles.
Notably, heteroaldehydes
27 were successfully used as reagents in chemosensors synthesis to detect cyanide ions (CN
−)
[11][12][33,34]; for example, indolium salts (hemicyanine derivatives)
28 were synthesized and used as colorimetric probes for CN
− recognition with limits of detection (LODs) of up to 0.99 μM
[12][34]. On the other hand, the 1-(2-pyridyl)-4-styrylpyrazole
29, obtained from
27 via a Witting olefination followed by a Mizoroki-Heck coupling, was used to detect Hg
2+ with a LOD of 0.31 μM
[13][35] (
Scheme 5Scheme 6b); these LODs values are below the respective limits of the World Health Organization (WHO)
[14][21].
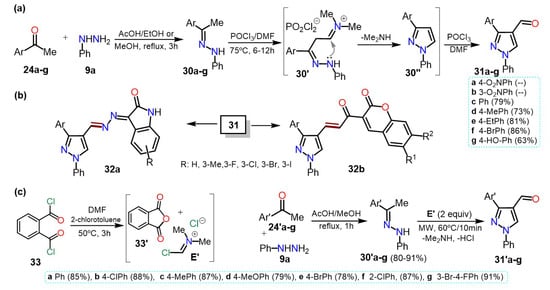
Similarly, Kaur et al.
[15][36] synthesized 4-formyl-1-phenylpyrazoles
31a–f using the Vilsmeier-Haack reaction with phenylhydrazones
30a–f, POCl
3, and DMF. The corresponding substrates
30a–f were obtained by a condensation reaction between phenylhydrazine (
9a) and acetophenones
24a–f in ethanol using acetic acid as a catalyst (
Scheme 6Scheme 7a).
HerIn this case
in, the yields are not particularly dependent on the different substituents.
Scheme 67.
Synthesis of (
a
) 4-formylpyrazoles and (
b
) biologically activity pyrazoles. (
c
) Reaction de VH using PDC
33
.
From 4-formylpyrazoles
31, new hybrid isatin derivatives
32a were obtained and tested for their α-Glucosidase inhibition for controlling postprandial hyperglycemia in diabetic patients. A similar methodology was used by Kumar and co-workers, who synthesized the 4-formylpyrazoles
31c–g starting from acetophenones
24c–g and
9a in yields between 63–86% (
Scheme 6Scheme 7b)
[16][37]. The heteroaldehydes
31c–g were used as intermediates for synthesizing pyrazole-coumarin derivatives
32b, and their antitubercular activity against the Mycobacterium tuberculosis H37Rv strain was tested.
Most formilations over pyrazole rings are carried out via a classical Vislmeier-Haack (VH) reaction (i.e., POCl
3/DMF), and modifications changing the chlorination agent can be performed so as not to use the toxic reagent POCl
3. For instance, Kumari et al.
[17][38] synthesized 4-formylpyrazoles
31′a–g in a similar route that shown in
Scheme 7a, that is, through the hydrazone derivative
30′a–g; nevertheless, the VH reagent was derived from phthaloyl dichloride (PDC,
33) and DMF (
SScheme 7c
heme 7c). Once the electrophile
E′ is formed, the reaction of it with
30′a–g is performed under MW irradiation to afford products in high yields (78–81%), without particular dependence on the effect of the substituent.
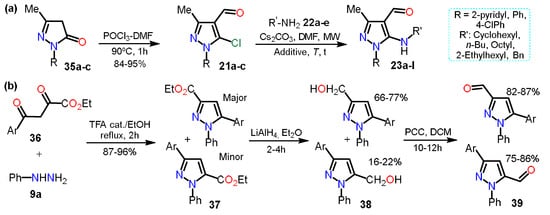
In the previous section, 5-alkylaminopyrazoles
23 were mentioned (see
Scheme 5b), where the substrates used for that synthesis were 5-chloro-4-formylpyrazoles
21, which were obtained from the respective pyrazolones
35; these starting materials undergo a chloroformylation reaction under Vilsmeier-Haack conditions to afford heteroaldehydes
21 (
Scheme 7Scheme 8a). In the next synthetic step, chlorine was substituted, generating only the 5-amino-4-formylpyrazoles
23 chemoselectively
[10][32].
Scheme 78.
Synthesis of formylpyrazoles from (
a
) pyrazolones and (
b
) diketoesters.
In practically all the literature about formylpyrazoles synthesis, the Vilsmeier-Haack conditions are used; however, in 2007, Nag et al. obtained 3/5-formyl derivatives
39 in an interesting and unconventional example
[18][19][39,40] that w
ase decided to consider since
it is we found no more examples of this methodology. For synthesizing products, pyrazole esters
37 were obtained by the cyclocondensation reaction between diketoesters
36a–c and phenylhydrazine (
9a). Subsequently, compounds
37 were reduced with LiAlH
4 in dry diethyl ether to give the respective pyrazole alcohols
38, which by PCC-promoted oxidation reaction yielded the desire 3/5-formylpyrazoles
39 in high yields (
Scheme 7Scheme 8b).
2.2. Other Acylated Derivatives
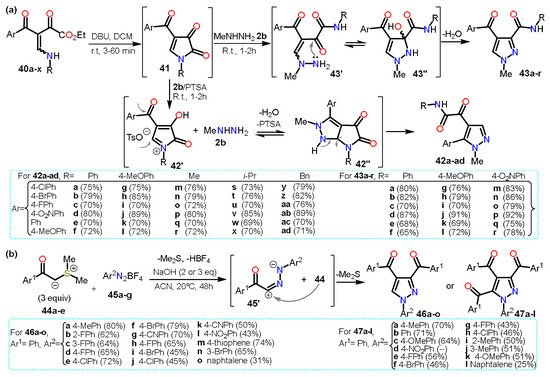
Regarding other acylpyrazoles, Poletto et al.
[20][41] recently developed a regioselective synthesis of 4,5/3,4-disubstituted
N-methylpyrazoles
42/
43 from 4-acyl-1
H-pyrrole-2,3-diones
41 and methylhydrazine
2b in the presence or not of acid (
Scheme 8Scheme 9a).
Scheme 89.
Synthesis of acylpyrazoles from (
a
) β-enaminodiketones and (
b
) sulfur ylides.
The pyrrole derivative 41 is generated in situ when the β-enaminodiketone 40a–x is cycled in the presence of DBU. Treatment of the pyrrole-2,3-dione 41 with p-toluenesulfonic acid (PTSA) leads to the formation of specie N-acyliminium 42′, which is then converted to the fused system pyrrolo[2,3-c]pyrazole 42″; finally, the cleavage of 42″ affords the 4,5-disubstituted pyrazoles 42a–ad. The absence of PTSA in the reaction allows 2b to directly attack C5 of the intermediate 41 followed by cleavage of the pyrrole ring generating a non-cyclic intermediate 43′. Afterward, an amino group performs a nucleophilic attack on the carbonyl carbon of the α-ketoamide group; ultimately, water elimination in 43″ gives the 3,4-disubstituted pyrazoles 43a–r. In both cases, high yields were obtained regardless of the substituents used, and various ERGs and EWGs were tested to evaluate the scope of the reaction.
Similarly, Qui et al.
[21][42] reported a divergent domino annulation reaction between sulfur ylides
44a–e with aryldiazonium tetrafluoroborates
45a–g to afford tri- and tetra-substituted acylpyrazoles
46a–o and
47a–l, respectively; this synthesis proceeded via the interaction of the in situ generated 1,3-dipole
45′ with more molecules of
44 (
Scheme 8Scheme 9b).
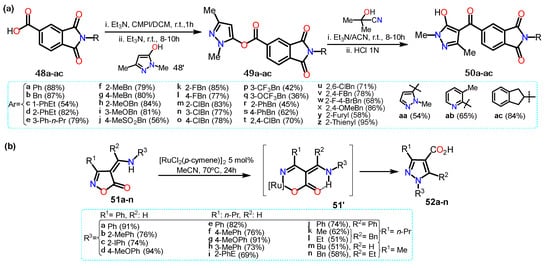
In a different work, He et al.
[22][43] synthesized acylpyrazoles
50a–ac from N-substituted isoindoline-1,3-dione derivatives
48a–ac (
Scheme 9Scheme 10a). Precursors
48a–ac were obtained by reaction between 1,3-dioxo-1,3-dihydroisobenzofuran-5-carboxylic acid with the appropriate primary amine in anhydrous acetic acid. The substrate
48a–ac and 2-chloro-1-methylpyridinium iodide (CMPI) reacted to then formed the respective pyrazole esters
49a–ac with 1,3-dimethyl-1
H-pyrazol-5-ol (
48′). The esters molecules were then transformed, through a Fries rearrangement, into the final products
50a–ac; these 4-aroylpyrazoles
50a–ac were tested for
Arabidopsis thaliana 4-hydroxyphenylpyruvate dioxygenase (AtHPPD) inhibition activities. For these derivatives, once EWGs were inserted the yields were slowly lower than for those with ERGs.
Scheme 910.
Synthesis of acylpyrazoles from (
a
) 1,3-dimethyl-1
H
-pyrazol-5-ol and (
b
) isoxazolones.
Recently, Loro et al.
[23][44] obtained pyrazole-4-carboxylic acids
52a–n starting from isoxazole-5(4H)-ones (
51a–n) using [RuCl
2(p-cymene)]
2 as a catalyst. The transformation begins with a ring-opening non-decarboxylative path that generates a vinyl Ru-nitrenoid intermediate that undergoes cyclization to afford the desired pyrazoles (
Scheme 9Scheme 10b). Specifically, the catalytic cycle starts with the oxidative addition of catalyst to
51, generating intermediate
51′, which is stabilized due to the formation of a hydrogen bonding; this complex undergoes ring-opening resulting in a Ru-nitrenoid intermediate affording the final product via reductive elimination of the metal; it is worth mentioning that the catalytic cycle mechanism is not well elucidated, and the authors explain just a proposal.
3. Further Functional Pyrazoles
3.1. Halopyrazoles
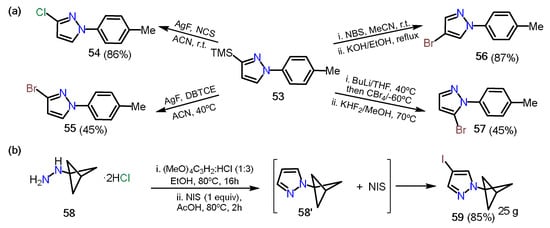
In 2019, Onodera et al.
[24][45] reported a regioselective halogenation of 3-trimethylsilylpyrazole
53 (
Scheme 10Scheme 11a). The introduction of halogen atoms at positions 3, 4, and 5 was possible thanks to the different character and orthogonal reactivity of each one; position 3 has the trimethylsilyl group (TMS), which can be easily removed under mild conditions generating a carbanion that can react towards electrophilic substrates such as
N-chlorosuccinimide (NCS) and 1,2-dibromotetrachloroethane (DBTCE), affording chlorinated
54 and brominated
55 pyrazoles, respectively. On the other hand, position 4 is the most nucleophilic on the ring; therefore, the direct reaction with
N-bromosuccinimide (NBS) followed by deprotection of the TMS group affords the 1-aryl-4-Bromopyrazole
56. Finally, position 5 possesses the most acidic proton of the ring; thus, using a base such as
n-butyllithium and tetrabromomethane, followed by deprotection of TMS, allows a halogenation at position 5 of the pyrazole ring to afford the respective 5-bromopyrazole
57. A similar approach was recently reported by Zarate and co-workers
[25][46], in which the authors synthesized the 4-iodopyrazole derivative
59 through a condensation/iodination sequence starting from bicyclo[1.1.1]pentan-1-ylhydrazine
58 and using tetramethoxypropane as an additive in the reaction carried out in ethanol (
Scheme 10Scheme 11b).
Scheme 101.
(
a
) Halogenation of the pyrazole
53
and (
b
) synthesis of the 4-iodopyrazole
59
.
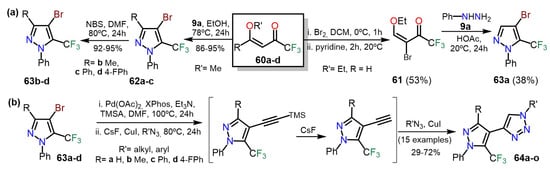
In 2017, Bonacorso et al.
[26][47] Synthesis of 4-bromo-5-(trifluoromethyl)-1-phenyl-1
H-pyrazoles
63a–d by two interesting methodologies; the first proceeded through the brominated 1,3-bis-electrophilic substrate
61 whereas, in the second, the pyrazole ring in
62 was brominated using NBS as the brominated agent. The synthesis of
63a–d was developed by utilizing 1,1,1-trifluoro-4-methoxy-alken-2-ones
60a–d as starting reagents. On the one hand, substrate
60a was brominated and then cyclocondensed with phenylhydrazine (
9a) to form product
63a. On the other hand,
60b–d cyclocondensed with
9a to obtain pyrazoles
62a–c, which finally brominated to obtain products
63b–d (
Scheme 11Scheme 12a). Compounds
63a–d were successfully used in the one-pot three-step synthesis of polysubstituted 4-(5-(trifluoromethyl)-1
H-pyrazol-4-yl)-1
H-1,2,3-triazoles
64a–o; they carried out a sequential Sonogashira cross-coupling, desilylation, and a copper(I)-catalyzed azide-alkyne cycloaddition reaction (CuAAC) with high overall yields. The authors cited that the CF
3 group in
63 made the Sonogashira cross-coupling reaction challenging (
Scheme 11Scheme 12b).
Scheme 112.
Synthesis of (
a
) 4-bromo-5-(trifluoromethyl)pyrazoles and their (
b
) synthetical utility.
As
itwe c
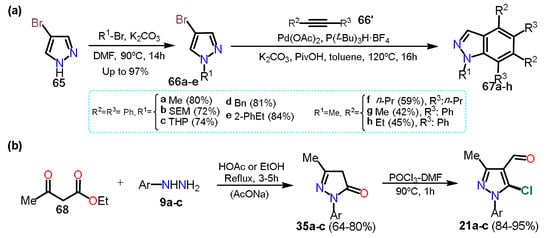
ould be seenan see, in the above approaches, halogenation of the pyrazole is carried out once the ring; however, other methods use the commercial halogenated pyrazole as a start reagent in the synthesis of more complex structures; these protocols exist due to the versatility of halosubstituted products, allowing different reactions such as aromatic substitutions on the rings or coupling reactions to form new C–C bonds. For example, Tsui and collaborators
[27][48] used 4-bromopyrazoles
66a–e in palladium-catalyzed benzannulation to obtain substituted indazoles
67a–h. The presence of bromine facilitates the oxidative addition step on C4; it is important to note that despite some halopyrazoles being commercial, various halogenated substrates are obtained by other transformations that do not involve direct halogenation; in this respect, the Tsui group synthesized the
N-alkyl-4-bromopyrazole derivative
66e by the respective
N-alkylation reaction of 4-bromo-1
H-pyrazole (
65) and alky bromides potassium carbonate-mediated in DMF (
Scheme 12Scheme 13a).
Scheme 123.
Synthesis of (
a
) indazoles from 4-bromopyrazoles and of (
b
) 5-chloropyrazoles.
In the above sections,
we cited the previous work of Orrego-Hernandez et al.
[10] was cited[32], in which 5-alkylaminopyrazoles
23 were obtained through NAS reactions on 5-chloro-4-formylpyrazoles
21 and using primary alkyl amines as nucleophiles. The substrates
21 were obtained by the chloroformylation reaction under Vilsmeier-Haack conditions of the respective pyrazolones
35 (see
Scheme 5b and
Scheme 8a). Consequently, this methodology is another fundamental example of access to halopyrazoles, particularly 5-chloropyrazoles, from ethyl acetoacetate (
68) as the starting material (
Scheme 12Scheme 13b).
3.2. Additional Systems
Throughout the entire contribution, several functionalized pyrazole derivatives have been mentioned (i.e., rings substituted with NH2, CHO, OH, CF3, SR, CN, CO2R, Cl, Br, etc.), and some of them managed to be classified within a particular section due to their recurrence (pyrazoles bearing amino or acyl groups); however, there are examples on other functional pyrazoles that are not part of such sections; therefore, in the last section of this chapter, seven works on different or highly functionalized pyrazoles are discussed.
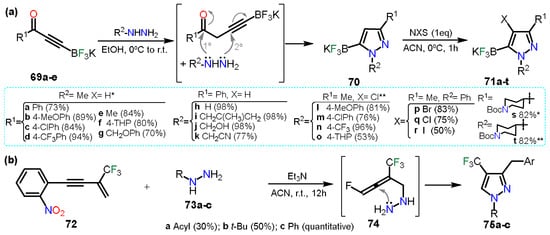
In the first example, Fricero et al.
[28][49] reported the regioselective condensation between ynone-trifluoroborates
69a–e and hydrazine derivatives to obtain pyrazole 5-trifluoroborates
70 (
Scheme 13Scheme 14a). The reaction generates a nitrile intermediate just such as the ones studied
in Section 2.1.
In Hereinthis case, products are stable, allowing a chemoselective halogenation of
70 to obtain the fully functionalized pyrazoles
71a–t. The halogenation methodology is such as the ones mentioned in the above section, using N-halosuccinimides and shows that the halogenation is compatible with the trifluoroborate systems as it does not undergo halodeborylations.
Scheme 134.
Synthesis of pyrazoles from (
a
) ynone trifluoroborates and (
b
) from 1,3-enynes.
On the other hand, Wei and co-workers
[29][50] reported the synthesis of trifluoromethylated pyrazoles
75a–c; these pyrazoles were obtained via a double hydroamination reaction of β-CF
3-1,3-enyne
72 with hydrazine derivative
73a–c (
Scheme 13Scheme 14b). First, reagents
72 and
73 undergo an intermolecular hydroamination generating intermediate
74, in which amine performs a nucleophilic attack over the central sp-carbon to obtain the cyclization products; it is to notice that product
75a was obtained alongside pyrazolidine which is the non-aromatized product. When
73c was used, pyrazolidine was obtained, but as it was air sensitive, it was readily oxidized into pyrazole
75c.
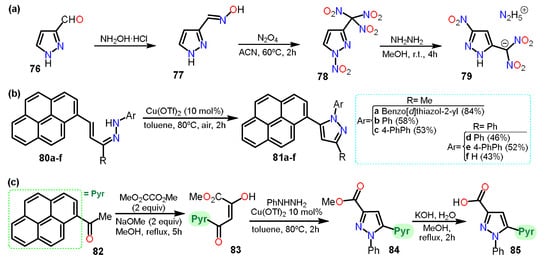
Another example is the preparation of nitro-substituted pyrazoles. Zhang et al.
[30][51] synthesized 1-nitro-3-trinitromethylpyrazole (
78) from 3-formylpyrazole (
76) (
Scheme 14Scheme 15a). Compound
78 was used to obtain hydrazinium 5-nitro-3-dinitromethyl-2
H-pyrazole
79. The synthetical route,
76 was treated with hydroxylamine hydrochloride to yield compound
77. Subsequently,
77 was treated with N
2O
4 to obtain
78, which reacted with hydrazine to obtain the dinitromethylide salt
79.
HerIn this case
in, in the process of dinitration, C5 was nitrated, too, making it clear that isomerization of
N-nitropyrazole was carried out during the last step. The isomerization mechanism was elucidated using DFT computational calculations, and the final product was used as an energetic salt.
Scheme 145.
(
a
) Synthesis of nitro pyrazoles and of pyrene-pyrazoles (
b
)
81
and (
c
)
85
.
Continuing, Sar et al.
[31][52] reported the synthesis of seven pyrene-pyrazole pharmacophores for targeting microtubules (
Scheme 14Scheme 15b,c). The pyrenyl-substituted pyrazoles
81a–f were prepared with the corresponding hydrazones
80a–f and had side-chain modifications at N-1 and C-3 positions, inserted from the alkenyl hydrazones via C-N dehydrogenative cross-coupling using a copper triflate catalyst under aerobic conditions. Furthermore, the reaction of pyrenylacetophenone (
82) with dimethyloxalate produced molecule
83 that then undergoes cyclization reaction with phenylhydrazine to produce
84 via C-N bond formation in one pot. Finally,
84 was treated with KOH/MeOH to yield
85.
The following two examples imply the amino or the keto group, and their preparation involves analog functional groups to the ones mentioned in the previous sections; though,
itwe is mention
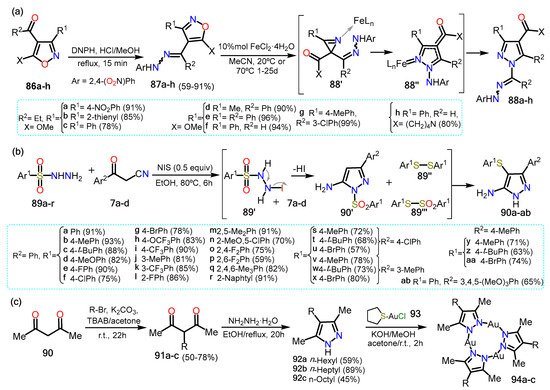
ed them since they are highly functionalized compounds. In this context, Galenko et al.
[32][53] synthesized 1-aminopyrazole-4-carboxylic acids using an iron II catalyst (
Scheme 15Scheme 16a). The synthesis starts with the isoxazoles
86a–h, which react with 2,4-dinitrophenylhydrazine (DNPH) to generate both
E/
Z isomers of 4-hydrazonomethylisoxazoles
87a–h. Afterward, the catalyst FeCl
2·H
2O is added with dry acetonitrile developing a domino rearrangement of the isoxazole via the formation of aziridine intermediate
87′. The mechanism starts with forming a Fe-isoxazole complex, followed by the ring’s opening via N–O bond cleavage to form a Fe-nitrene complex; this complex then undergoes recyclization to form the Fe-azirine complex
87′. The three-membered ring is open, generating another Fe-nitrene complex that allows the 1,5-cyclization producing the complex Fe-
N-aminopyrazole
87″. Lastly, cleavage of the catalyst affords pyrazoles
88a–h in high yields. Notably, the yields shown are obtained starting from the
E isomer of the isoxazoles, although the reaction proceeds smoothly for both isomers.
Scheme 156.
Synthesis of (
a
) pyrazoles
88
, (
b
) aminopyrazoles
90
and (
c
) trinuclear complexes
94
.
Later, Wei et al.
[33][54] reported a three-component reaction of aroylacetonitriles
7a–x with arylsulfonyl hydrazides
89a–r to form 5-amino-4-arylthio-3-aryl-1
H-pyrazoles (
Scheme 15Scheme 16b). The reaction could afford 1-
H or 1-SO
2Ph products, but in the presence of NIS, the reaction became selective to the 1
H-pyrazole. Various substituents in arylsulfonyl hydrazides and the β-ketonitrile were tested to investigate the scope of the reaction; it was found that the electronic effects of the aryl group did not influence the reaction. The mechanism reaction proceeds via sequential cyclization and sulfenylation reactions under NIS catalysis. The reaction starts with the reduction of
89′ that after losing HI and N
2, affording two disulfide spices; that is, 1,2-diphenyldisulfane
89″ and S-phenylbenzenesulfonothioate
89‴. Meanwhile,
7 reacts with
89 via a cyclization reaction to generate the desired 5-amino-1-arylthio-3-arylpyrazole
7′. Two routes are possible to afford the final products. In the first one,
7′ undergoes electrophilic substitution with
89 to afford the arylthio group at position 4; this product treated with NIS provides the desired aminopyrazoles
90a–ab. In the other proposed route,
7′ then loses the arylthio group in the presence of NIS and reacts with
89 to afford
90a–ab which possesses the thioether group at position 4
.To finish, Tsutsumi et al. [34] reported phosphorescent trinuclear Au(I) complexes using NH-pyrazoles as ligands (Scheme 15c). Pyrazole is prepared from 2,4-pentanedione (90), which is alkylated using K2CO3 as a base and an alkyl bromide to afford the α-substituted β-diketones 91a–c. Afterward, 91a–c undergoes a cyclization reaction with hydrazine to afford the pyrazoles 92a–c that then were used to prepare the trinuclear complexes 94a–c with tetrahydrothiophene-AuCl (93). Complexes 94 were recrystallized, and all the crystals exhibited broad unstructured luminescence around 730 nm with quantum yields of 75%, 61%, and 63%, respectively. The complexes did not reveal a good luminescence in diluted solutions; however, the isolated molecules exhibited the opposite behavior, indicating that the formation of aggregates induces the luminescence of the complexes.