Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Linya You and Version 3 by Sirius Huang.
The bromodomain and PHD finger–containing protein1 (BRPF1) is a member of family IV of the bromodomain-containing proteins that participate in the post-translational modification of histones. Patients with BRPF1 mutations often display intellectual disability or suffer from leukemia or medulloblastoma. BRPF1 is an activator and a scaffold protein of a multiunit complex, with other members being KAT6A/KAT6B, the inhibitor of growth 5 (ING5) or ING4 and MYST/Esa1-associated factor 6 (MEAF6)EAF6.
- BRPF1
- KAT6A
- KAT6B
- intellectual disability
- neurodevelopmental disorders
- cancer
- mutations
1. Neurodevelopmental Disorders Associated with Mutations in BRPF1/KAT6A/KAT6B
Fish and mouse BRPF1-related studies have demonstrated that BRPF1 has essential roles in embryo development, forebrain development, hematopoiesis, skeletal patterning and synaptic transmission. Thus, an interesting question is whether BRPF1 mutations in humans cause developmental abnormalities. To date, 43 cases of BRPF1 mutations reported confirm that BRPF1 is a causal gene for intellectual disability (ID) in a disease known as intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) (12 cases [1][13], 10 cases [2][106], 12 cases [3][107], 1 case [4][108], 1 case [5][109], 1 case [6][110], 4 cases [7][111], 1 case [8][112], 1 case with schizophrenia and mild ID [9][113]). The sites of BRPF1 mutations involved in IDDDFP are summarized in Figure 1.

Figure 1. Syndromic intellectual disability-associated BRPF1 germline variants. (A) Illustration of the BRPF1 variants identified in the 43 cases identified to date. A BRPF1 Tyr406His variant was identified in an autistic individual, but the pathogenicity remains elusive. (B) Lollipop graph demonstrating the distribution of syndromic intellectual disability-associated BRPF1 variants in different domains. Most variations are clustered in PZP domain.
Yan et al. [2][106] identified 10 individuals with 9 different mutations of the BRPF1 gene, all of whom displayed intellectual disability, global developmental delay, expressive language impairment and impaired H3K23 acetylation. Among the 9 BRPF1 variants, 7 were de novo mutations and 2 were inherited from their mothers. The missense mutation p.Pro370Ser is located within the PZP domain. The other 8 truncating mutations encode variants missing essential structural domains of BRPF1. The variants p.Glu121Glyfs*2, p.Trp315Leufs*26, p.Arg455* and p.His563Profs*8 lack the ING5- and MEAF6-interacting domain. By contrast, the remaining 4 variants p.Gln629Hisfs*34, p.Arg833*, p.Met973Asnfs*24 and p.Arg1100* have complete ING5- and MEAF6-interacting domain. Moreover, this team also analyzed these variants’ functional impact on the formation of tetrameric complexes, the acetyltransferase activity of KAT6A and subcellular localization. p.Pro370Ser, p.Gln629Hisfs*34, p.Arg833* and p.Arg1100* can promote production of ING5 and MEAF6 and form tetrameric complexes in HEK293 cells as wild-type BRPF1. However, p.Glu121Glyfs*2, p.Trp315Leufs*26 and p.Arg455* cannot promote ING5 and MEAF6 expression. Among them, p.Glu121Glyfs*2 failed to interact with KAT6A while the remaining 2 can interact with KAT6A. However, p.Arg455* failed to mediate the interaction of KAT6A with ING5 and MEAF6. Surprisingly, pTrp315Leufs*26 can still interact with MEAF6. For acetyltransferase activity, p.Pro370Ser, pTrp315Leufs*26 and p.Arg455* showed reduced stimulation of KAT6A activity, while p.Gln629Hisfs*34, p.Arg833* and p.Arg1100* were as active as wild-type BRPF1. At last, the variants behaved differently from wild-type BRPF1 in subcellular localization. p.Glu121Glyfs*2 and p.Trp315Leufs*26 presented uniform cytoplasmic distribution, p.Arg833* formed large aggregates in the cytoplasm and p.Arg455* and p.Gln629Hisfs*34 were mainly nuclear. In the presence of KAT6A, ING5 and MEAF6, these variants all became nuclear. Thus, the 9 variants appear to generate different groups, suggesting their deregulation of BRPF1 via distinct mechanisms.
Mattioli et al. [3][107] identified 12 individuals carrying 5 BRPF1 mutations, 1 nonsense and 4 splice variants. All individuals with BRPF1 mutations have mild or moderate ID. One variant was a 2 nt deletion, p.Val351Glyfs*8, which retains the KAT6A/KAT6B interaction domain but lacks the ING5-MEAF6 interaction domain, leading to failure of complex formation, failure of H3K23Ac stimulation and more uniform distribution in both cytoplasm and nucleus. The remaining 4 were mutations of the BRPF1 gene, 1 de novo missense variant—p.Cys389Arg and 3 nonsense or frameshift variations—p. Tyr994*, p.Asp190Metfs*24 and p.Tyr35*.
Yan et al. [1][13] recently reported another 12 cases of syndromic intellectual disability and demonstrated that these and previous cases also showed impaired H3K23 propionylation. Intellectual disability, language delay and facial/eye dysmorphisms (eg. blepharophimosis and ptosis) were frequently observed. 11 BRPF1 variants were identified in the 12 cases. They were p.Pro76Leu, p.Gln96*, p.Asp187Glyfs*29, p.Met295Valfs*17, p.Arg318His, p.His410Arg, p.Thr434Profs*61, p.Glu474Glyfs*3, p.Tyr543Thrfs*6, p.Arg833* and p.Phe1154del. p.Arg833* was previously reported and thus there were 10 new variants. 6 of them led to C-terminal truncations (Figure 1). p.Gln96* and p.Asp187Glyfs*29 variants lack the KAT6A/KAT6B-interacting domain. p.Met295Valfs*17 and p.Thr434Profs*61 variants lack a complete PZP domain, which is critical for BRPF1 to promote nucleosomal H3K23Ac. p.Glu474Glyfs*3 and p.Tyr543Thrfs*6 lack an intact EPC-II domain required for ING5/MEAF6 binding. Thus, the 6 variants are probably causative. For the remaining 4 variants, p.His410Arg possibly disrupts the PZP domain. p.Phe1154del likely inactivates the PWWP domain. p.Pro76Leu disrupts the N-terminal region, whereas p.Arg318His alters the first PHD of the PZP domain (Figure 1). Function-associated studies demonstrated that p.Arg318His can form a tetrameric complex normally, whereas p.Thr434Profs*61 could not interact with ING5 and MEAF6. The 2 variants were both defective for stimulating H3K23 acetylation and propionylation by KAT6A. Surprisingly, p.Pro76Leu was the exception with normal promotion of ING5 and MEAF6 expression and normal stimulation of H3K23 acylation by KAT6A as wild-type BRPF1. Thus, BRPF1 mutations appear to deregulate its functions through different mechanisms.
4 other de novo truncating variants (BRPF1-p.Gln629Hisfs*34, p.Val707Argfs*8, p.Arg833*, and p.Met973Asnfs*24) have also been identified in 4293 UK individuals in the Deciphering Developmental Disorders (DDD) study [10][114]. Additional BRPF1 variants reported include a de novo LoF variant (p.Ala396LeufsTer69) in a child of sudden unexplained death [8][112], a truncating variant (p.Q186*) in three affected siblings and their mother [7][111], a variant (p.Val352Leu) in a girl [6][110], a de novo nonsense variant (p.Glu219*) in a boy [5][109] and a rare nonsense variant (p.Gln322*) in a patient with normal intellectual development [4][108]. A BRPF1 Tyr406His variant was identified in an autistic individual, but the pathogenicity remains elusive [11][115].
In addition, BRPF1 was identified as the most clinically relevant genes required for dystonia by performing whole exome sequencing (WES)-based copy-number variation analysis [12][116]. Another study found that BRPF1 may be potentially disease-related for coloboma and microphthalmia [13][117]. BRPF1 is also one of the target genes regulated by pmiR-chr, which was significantly dysregulated in major depressive disorder patients [5][109].
KAT6A and KAT6B were originally identified as genes rearranged in leukemia [14][15][17,31]. Later, they were also reported to be mutated in patients with intellectual disability and neurodevelopmental disorders [16][17][18][19][20][21][22][23][24][36,37,118,119,120,121,122,123,124]. A recent study summarized 61 KAT6A variants from 76 patients [23][123]. Syndromes of 100% penetrance include intellectual disability and speech delay. The protein domains of KAT6A include a NEMM domain (aa 1-206), two PHD domains (aa 207-313), an MYST domain (aa 314-787), an acidic domain (aa 788-1414) and a Ser/Met domain (aa 1414-2004) (Figure 2A). The 61 variants were located spanning all domains (Figure 2A,C). Individuals with truncating mutations located in exons 16–17 of KAT6A showed more prevalent and severe ID.
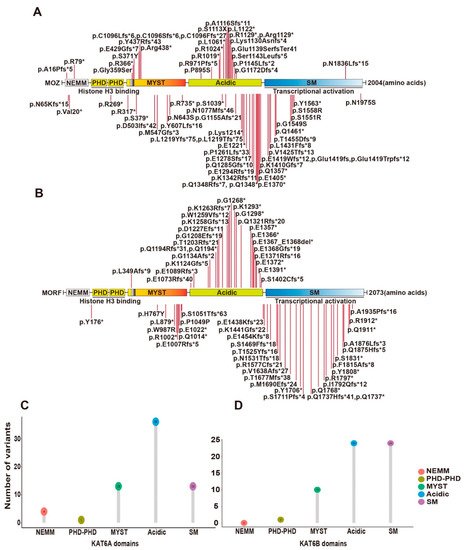
Figure 2. Syndromic intellectual disability-associated KAT6A and KAT6B germline variants. (A) Cartoon representation of KAT6A germline mutants identified in patients with intellectual disability. (B) Cartoon representation of KAT6B germline mutants identified in patients with intellectual disability. (C) Lollipop graph demonstrating the distribution of syndromic intellectual disability-associated KAT6A variants. Most variations are clustered in the acidic region. (D) Lollipop graph demonstrating the distribution of syndromic intellectual disability-associated KAT6B variants. Most variations are clustered in acidic and SM regions.
Other KAT6A variants reported since this report include a de novo frameshift variant (p.Lys1130Asnfs*4) in a 2-year-old boy with global developmental delay and ID [25][125], a de novo frameshift variant (p.Glu1419fs) in a 16-year-old girl with severe ID and pancraniosynostosis (no major visible skull suture lines) [26][126], 5 de novo variants (p.Gly359Ser, p.Arg1129*, p.Lys1214*, p.Ser1143Leufs*5, p.Glu1419Trpfs*12) from 5 patients with moderate or severe ID and severely affected speech and expressive language [27][127], a de novo variant (p.Glu1139SerfsTer41) in a 9-month-old boy with severe developmental delay [28][128], a variant (p.Arg438*) in a 2-month-old baby with multiple facial deformities [29][129], 2 novel variants (p.P1261Lfs*33) in a patient associated with pan-suture craniosynostosis [30][130], a missense variant (p.N1975S) in the index patient displaying microcephaly and developmental delay [31][131] and 2 de novo variants (p.S1113X [32][132] and p.Val20* [33][133]) in a 21-year-old man and a 1.2-year-old baby with intellectual disability, respectively (Figure 2A).
Mutations in KAT6B have been reported in patients with Say–Barber–Biesecker–Young–Simpson syndrome (SBBYSS or Ohdo syndrome) [19][119], genitopatellar syndrome (GPS) [20][21][120,121], and Blepharophimosis–Ptosis–Epicanthus inversus syndrome (BPES) [22][122]. Known cases with KAT6B variants have exceeded 60 with SBBYSS and GPS [24][124]. The two syndromes share features such as intellectual disability but also have their own particular symptoms, which seem to be dependent on the location of KAT6B mutations. SBBYSS-associated variants frequently appear in the distal part of exon 18, while GPS-associated variants are often distributed in the end of exon 17 and beginning of exon 18. The 60 known variants are summarized in Figure 2B,D.
2. Cancers Associated with the BRPF1-KAT6A/KAT6B Complex
In addition to germline mutations in patients with neurodevelopmental disorders, somatic mutations of BRPF1 have been reported in leukemia, medulloblastoma and other types of cancer [34][35][59,134] (Figure 3). About 236 BRPF1 variants have been found in 211 individuals out of a total of 10,240 cancer patients from TCGA datasets, equivalent to a prevalence rate of 2%. Furthermore, about 1016 cases with copy number variation (CNV) events of BRPF1 are found in 11,115 cancer patients, corresponding to a rate of 10%. Thus, BRPF1 is frequently mutated in different cancer types [1][13]. The impact of each cancer-derived somatic BRPF1 mutation should be verified experimentally. Mutants Pro20Leu, Arg29Cys and Ser36Ile alter the BRPF1-specific SZ domain, and affect complex formation and H3K23Ac. Mutants Arg66Cys, Arg59His, Arg59Cys and Gln67Pro likely affect NLS1 function, while mutants Glu253Gly, Leu298Pro, Trp348Arg and Glu369Asp, identified in medulloblastoma, are located in the EPC-I and PZP domains, respectively, and exert variable effects on enzyme activity.

Figure 3. Cancer-associated BRPF1 somatic mutants. (A) Cartoon illustration of somatic variants of BRPF1 identified in cancer. (B) Lollipop graph demonstrating the distribution of cancer-associated BRPF1 variants.
In addition to mutations, accumulating findings have indicated BRPF1’s role in cancer. Truncated BRPF1 protein, cooperating with SmoM2 activation, promotes postmitotic neuron dedifferentiation, re-entering the cell cycle and inducing medulloblastoma in vivo [36][135]. BRPF1, as an inflammatory signature gene in glioma, regulates glioma cell proliferation and colony formation, thereby being described as a potential drug target of primary lower-grade gliomas [37][136]. In addition, BRPF1 is significantly upregulated in human hepatocellular carcinoma [38][137] and was found to be a biomarker to discriminate prostate cancer patients and healthy controls [39][40][138,139].
Recent pan-cancer analysis of CNV has identified KAT6A and KAT6B as top targets for amplification in different cancers [41][140]. In humans, abnormal chromatin acetylation caused by KAT6A may be a contributing factor to cancer. KAT6A was reported to cooperate with TP53 to drive cancer growth [42][141]. Inhibition of KAT6A/KAT6B induces senescence and arrests tumor growth [43][142]. KAT6A was frequently reported to be translocated in various hematological malignancies to form fusion genes, such as KAT6A-CBP, KAT6A-TIF2 and KAT6A-EP300 [14][17]. Similarly, KAT6B is also rearranged in leukemia [44][32]. In addition to hematologic malignancies, recurrent amplifications of KAT6A have been reported in various solid tumors, including breast cancer, ovarian cancer, uterine cervix cancer, lung adenocarcinoma, colon and rectal cancer [45][143]. In addition, KAT6A and structurally similar gene KAT6B also undergo rearrangements in myelodysplastic syndromes [46][144] and benign uterine fibroids [44][32].
KAT7 is overexpressed in cancerous tissues [47][145]. Its substrate specificity of H4 lysine is similar to the pattern of H4 modification observed in cancer [48][11]. The KAT7 gene maps to 17q21.3, the region of which is associated with frequent allelic gains found in breast cancer and linked with poor prognosis [49][50][146,147]. In addition, KAT7 is essential to sustain functional leukemia stem cells [51][148], and its overexpression facilitates osteosarcoma [52][149] and hepatocellular carcinoma growth [53][150].
ING4 downregulation, loss of expression and mutations have been observed in many tumors and cancer cell lines, supporting its potential as a tumor suppressor that regulates several biological and pathological processes [54][151]. However, the loss of ING4 alone is not sufficient to trigger tumorigenesis [55][152], consistent with its interaction with signaling pathways such as MYC, TP53, NF-κB and HIF-1 in tumor suppressive functions [54][151]. ING4 dysregulation correlates with pathophysiological process of many tumors, such as astrocytomas [56][153], clear-cell renal carcinoma [57][154], glioblastoma [58][97], glioma [59][155] and hepatocellular carcinoma [60][156]. Similarly, ING5 manipulates tumor progression via interaction with different molecules [61][157]. Nuclear ING5 is negatively correlated with tumor size and depth of invasion [62][158], while cytoplasmic ING5 is associated with tumor progression [63][159].
3. Other Diseases Associated with BRD1 and BRPF3
Inactivation of BRD1 in mice led to lethality of E15.5 embryos with growth retardation, neural tube defects, abnormal eye development and erythropoiesis [64][81]. BRD1 also regulates embryogenesis and early thymocyte development [64][65][81,160]. In humans, PAX5-BRD1 fusion events have been reported in leukemia [66][161]. BRD1 is also associated with bipolar disorder and schizophrenia in European populations [67][162].
Endogenous BRPF3 preferentially forms tetrameric complexes with KAT7, and it is not essential for mouse embryo survival, distinguishing it from its homologs BRPF1 and BRD1 [68][163]. Others reported that BRPF3 is essential for DNA replication initiation and damage response in immortalized cell lines [69][82]. Few have reported BRPF3 mutation events in humans.