The lung epithelium is constantly exposed to harmful agents present in the air that we breathe making it highly susceptible to damage. However, in instances of injury to the lung, it exhibits a remarkable capacity to regenerate injured tissue thanks to the presence of distinct stem and progenitor cell populations along the airway and alveolar epithelium. Mechanisms of repair are affected in chronic lung diseases such as idiopathic pulmonary fibrosis (IPF), a progressive life-threatening disorder characterized by the loss of alveolar structures, wherein excessive deposition of extracellular matrix components cause the distortion of tissue architecture that limits lung function and impairs tissue repair. cHere, we reviell therapy has been investigated for the treatment of IPF, including the use of a variety of cell types such as lung epithelial cells, specifically AT2 cells, induced pluripotent stem cells (iPSCs), and mesenchymal stem cells (MSCs) isolated from bone marrow stroma and those from adipose tissue or from other tissuesw the most recent findings of a study of epithelial cells with progenitor behavior that contribute to tissue repair as well as the mechanisms involved in mouse and human lung regeneration. In addition, we describe therapeutic strategies to promote or induce lung regeneration and the cell-based strategies tested in clinical trials for the treatment of IPF. Finally, we discuss the challenges, concerns and limitations of applying these therapies of cell transplantation in IPF patients. Further research is still required to develop successful strategies focused on cell-based therapies to promote lung regeneration to restore lung architecture and function.
- alveolar epithelial cells
- cell therapy
- lung regeneration
- pulmonary fibrosis
- stem/progenitor cells
- tissue homeostasis
1. Introduction
1.1. The Alveolar Compartment and the Development of Pathological Fibrosis
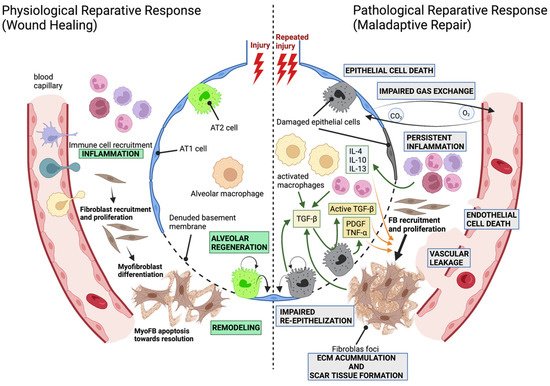
1.2. Modeling Lung Fibrosis

2. Regeneration and Stem/Progenitor Cells
2.1. Epithelial Stem and Progenitor Cells Are Contributing to Regenerate the Alveolar Epithelium
The adult lung epithelium is highly susceptible to damage since, due to its major function of gas exchange, it is constantly exposed to insults from the external environment including toxic chemicals and pathogens—virus or bacteria—which makes the lung vulnerable. However, although the lung is relatively quiescent at homeostasis, it shows a significant regenerative capacity in response to injury. This remarkable ability to repair damaged tissue is due to the presence of multiple stem and progenitor cell populations that quickly respond to injury (Figure 2) [47][48][49][50][51][52][66,67,68,69,70,71].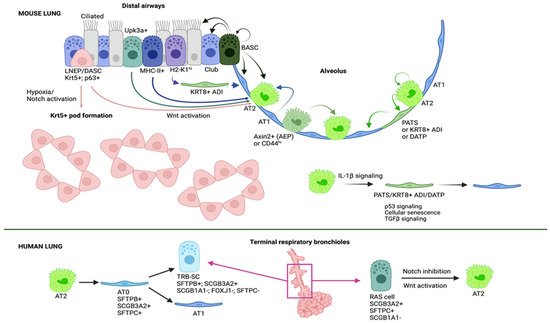 Figure 2. Distal airway/alveolar progenitors contributing to lung regeneration. A myriad of cells serve as alveolar progenitors upon damage. AT2 cells can proliferate and differentiate into AT1 cells after going through an intermediate cell state named pre-alveolar type-1 transitional cell state (PATS), keratin 8-positive alveolar differentiation intermediate (Krt8+ADI) or damage associated transient progenitors (DATPs). IL-1β produced by interstitial macrophages promotes AT2 differentiation into this intermediate cell type that displays a transcriptional signature of p53 signaling, cellular senescence and TGFβ signaling. Alveolar epithelial progenitors (AEPs) represent a subset of AT2 cells characterized by the expression of Axin2 that act as the principal progenitor cell population during injury-induced alveolar regeneration. CD44hi-expressing AT2 cells show an increased proliferative capacity also contributing to the regeneration of the alveolar epithelium. Interestingly although rarely, AT1 cells are able to dedifferentiate and give rise to AT2 cells. Bronchoalveolar stem cells (BASCs) are cells contributing to both alveolar and airway regeneration because of their ability to self-renew and to give rise to AT2, AT1, club and ciliated cells. Furthermore, subsets of club cells such as Upk3a+ subset, H2-K1hi subset and MHC-II+ subset can differentiate into AT2 cells; the latter go through a transitional state similar to KRT8+ ADI cells to give rise to AT1 cells. Of note, H2-K1hi and MHC-II+ club cells show an identical transcriptional signature suggesting that they are the same subpopulation. A rare population of p63+ cells in terminal bronchioles have shown the ability to activate Krt5 expression and expand and migrate to sites of injury. There, these cells give rise to AT2 cells or form pod-like metaplastic structures in a process regulated by hypoxic conditions, Notch signaling and Wnt signaling. Studies in human and non-human primate models have identified two interesting cell populations: AT0 cells, a novel bi-potential transient state that arises in the differentiation from AT2 cells into either terminal respiratory bronchiole secretory cells (TRB-SCs) or AT1 cells; and RAS cells, an airway secretory cell population that can differentiate into AT2 cells in a process regulated by Notch and Wnt signaling (Image created with BioRender).
In recent years, the advent of technologies such as single cell RNA sequencing (scRNASeq), spatial transcriptomics and advanced imaging has allowed to identify novel cell types of the lung and subpopulations, as well as transcriptional patterns and dynamics in healthy and diseased tissues. Multiple studies using single cell transcriptomics in combination with lineage tracing, lung injury models and organoid cultures have revealed that depending on the site of the damage (airway vs. alveoli) as well as the type and the extent of injury, different epithelial progenitor populations with the capacity to self-renew and differentiate into the epithelial lineages of the tissue respond to restore tissue homeostasis [48][50][53][54][67,69,72,73].
In the alveolar compartment, lineage tracing experiments have demonstrated that AT2 cells serve as progenitor cells of the homeostatic adult alveolar epithelium that are able to proliferate and differentiate into AT1 cells [55][56][57][58][59][60][74,75,76,77,78,79]. AT2 cells are mostly quiescent in the steady-state lung, but during repair after influenza infection, bleomycin exposure, hyperoxia, pneumonectomy-induced injury and genetic ablation of alveolar epithelial cells, AT2 cells quickly re-enter the cell cycle and produce new alveolar epithelial cells to keep the integrity of the alveolar epithelium and repair damage [47][52][59][60][61][62][63][64][65][66][66,71,78,79,80,81,82,83,84,85].
Although initially AT2 cells were thought to be a homogeneous population, recent studies have identified different subpopulations with higher regenerative capacities [67][68][69][86,87,88]. A subset of AT2 cells characterized by the expression of Axin2, a transcriptional target of Wnt signaling, and termed alveolar epithelial progenitors (AEPs) [69][88] are found at sites of severe damage and serve as the principal progenitor cell population during injury-induced alveolar regeneration [68][69][87,88]. Axin2+ AT2 cells were identified by two independent studies, both of them showing that these cells are highly proliferative and can further differentiate into AT1 cells following bleomycin- and influenza virus-induced lung injury [52][68][69][71,87,88]. The proportion of Axin2-expressing AT2 cells significantly varied between these two studies likely due to the use of different experimental strategies, but both demonstrated the expansion of this subpopulation after injury and their contribution to alveolar repair [68][69][87,88]. Another subset of AT2 cells is characterized by the expression of CD44 and exhibit high rates of proliferation suggesting a role in alveolar regeneration [67][86]. Whether Axin2+ and CD44+ AT2 cells are the same or a different subpopulation is still to be confirmed.
In addition to AT2 cells, considered as primary contributors to post-injury alveolar regeneration, various epithelial progenitor populations have been shown to participate in regeneration after damage [58][59][70][77,78,89].
BASCs (Bronchoalveolar Stem Cells) are found at the bronchoalveolar duct junction and they have been long understood to be stem cells that give rise to both AT2 cells and Club cells [71][72][90,91]. They were first defined by their anatomical location and the co-expression of the club cell marker, SCGB1A1, and the AT2 cell marker, SFTPC, suggesting their potential to differentiate into both cell types under a condition of lung injury. It was not until recently that it has been demonstrated, using two different approaches to lineage trace these cells in vivo, that indeed BASCs serve as progenitors of the distal airway and alveolar epithelium, being able to generate club cells, ciliated cells, AT2 and AT1 cells, confirming their contribution to lung regeneration [73][74][75][92,93,94].
Besides BASCs, airway epithelial stem/progenitor cell populations have been shown to contribute to both airway and alveolar regeneration [70][71][72][74][76][77][78][79][80][81][82][83][84][85][86][87][88][89,90,91,93,95,96,97,98,99,100,101,102,103,104,105,106,107]. Among these populations, several subsets of club cells in the terminal bronchioles, including Upk3a+ variant club cells, H2-K1 high club cells and MHC-II+ club cells, have been identified to give rise to AT2 cells in different models of severe damage [64][76][78][79][85][83,95,97,98,104].
A rare population of p63-expressing cells was identified in terminal bronchioles and their contribution to airway and alveolar regeneration was examined [80][82][83][86][99,101,102,105]. After severe injury such as bleomycin exposure or influenza infection, this rare population of the airway (Lineage Negative Epithelial Progenitors (LNEPs) [82][101]; Distal Airway Stem Cells (DASCs) [80][99]) activate the expression of Krt5, expand and migrate to damaged regions of the alveoli. However, these cells rarely give rise to AT2 cells nor to AT1 cells [82][86][101,105], but instead form pod-like metaplastic structures in which the alveoli are not repaired which persist for a long time and are likely thought to fill the injured space to preserve structural integrity. Nevertheless, Xi et al. demonstrated using stringent lineage tracing tools that hypoxia and Notch and Wnt signaling regulate the fate decision of p63+ progenitors to either regenerate the alveolar epithelium or to form these pod-like structures [70][89].
Surprisingly, recent evidence demonstrates that even terminally differentiated AT1 cells are able to dedifferentiate and give rise to AT2 cells after pneumonectomy and hyperoxia-induced lung injury [89][90][108,109], reflecting the significant plasticity of lung cells and conferring the lung as a valuable mechanism of repair. A tonic activation of YAP/TAZ is required to maintain AT1 identity, and their loss induces their differentiation into AT2 cells [90][109]. This is of high interest since it reveals mechanisms of the active maintenance of quiescence [48][67] necessary to keep tissue homeostasis similar to the Notch2 tonic activation in Club cells [91][110] that has been previously reported and that is required to maintain their cell identity and preserve adequate proportions of each cell type in the airway epithelium.
Figure 2. Distal airway/alveolar progenitors contributing to lung regeneration. A myriad of cells serve as alveolar progenitors upon damage. AT2 cells can proliferate and differentiate into AT1 cells after going through an intermediate cell state named pre-alveolar type-1 transitional cell state (PATS), keratin 8-positive alveolar differentiation intermediate (Krt8+ADI) or damage associated transient progenitors (DATPs). IL-1β produced by interstitial macrophages promotes AT2 differentiation into this intermediate cell type that displays a transcriptional signature of p53 signaling, cellular senescence and TGFβ signaling. Alveolar epithelial progenitors (AEPs) represent a subset of AT2 cells characterized by the expression of Axin2 that act as the principal progenitor cell population during injury-induced alveolar regeneration. CD44hi-expressing AT2 cells show an increased proliferative capacity also contributing to the regeneration of the alveolar epithelium. Interestingly although rarely, AT1 cells are able to dedifferentiate and give rise to AT2 cells. Bronchoalveolar stem cells (BASCs) are cells contributing to both alveolar and airway regeneration because of their ability to self-renew and to give rise to AT2, AT1, club and ciliated cells. Furthermore, subsets of club cells such as Upk3a+ subset, H2-K1hi subset and MHC-II+ subset can differentiate into AT2 cells; the latter go through a transitional state similar to KRT8+ ADI cells to give rise to AT1 cells. Of note, H2-K1hi and MHC-II+ club cells show an identical transcriptional signature suggesting that they are the same subpopulation. A rare population of p63+ cells in terminal bronchioles have shown the ability to activate Krt5 expression and expand and migrate to sites of injury. There, these cells give rise to AT2 cells or form pod-like metaplastic structures in a process regulated by hypoxic conditions, Notch signaling and Wnt signaling. Studies in human and non-human primate models have identified two interesting cell populations: AT0 cells, a novel bi-potential transient state that arises in the differentiation from AT2 cells into either terminal respiratory bronchiole secretory cells (TRB-SCs) or AT1 cells; and RAS cells, an airway secretory cell population that can differentiate into AT2 cells in a process regulated by Notch and Wnt signaling (Image created with BioRender).
In recent years, the advent of technologies such as single cell RNA sequencing (scRNASeq), spatial transcriptomics and advanced imaging has allowed to identify novel cell types of the lung and subpopulations, as well as transcriptional patterns and dynamics in healthy and diseased tissues. Multiple studies using single cell transcriptomics in combination with lineage tracing, lung injury models and organoid cultures have revealed that depending on the site of the damage (airway vs. alveoli) as well as the type and the extent of injury, different epithelial progenitor populations with the capacity to self-renew and differentiate into the epithelial lineages of the tissue respond to restore tissue homeostasis [48][50][53][54][67,69,72,73].
In the alveolar compartment, lineage tracing experiments have demonstrated that AT2 cells serve as progenitor cells of the homeostatic adult alveolar epithelium that are able to proliferate and differentiate into AT1 cells [55][56][57][58][59][60][74,75,76,77,78,79]. AT2 cells are mostly quiescent in the steady-state lung, but during repair after influenza infection, bleomycin exposure, hyperoxia, pneumonectomy-induced injury and genetic ablation of alveolar epithelial cells, AT2 cells quickly re-enter the cell cycle and produce new alveolar epithelial cells to keep the integrity of the alveolar epithelium and repair damage [47][52][59][60][61][62][63][64][65][66][66,71,78,79,80,81,82,83,84,85].
Although initially AT2 cells were thought to be a homogeneous population, recent studies have identified different subpopulations with higher regenerative capacities [67][68][69][86,87,88]. A subset of AT2 cells characterized by the expression of Axin2, a transcriptional target of Wnt signaling, and termed alveolar epithelial progenitors (AEPs) [69][88] are found at sites of severe damage and serve as the principal progenitor cell population during injury-induced alveolar regeneration [68][69][87,88]. Axin2+ AT2 cells were identified by two independent studies, both of them showing that these cells are highly proliferative and can further differentiate into AT1 cells following bleomycin- and influenza virus-induced lung injury [52][68][69][71,87,88]. The proportion of Axin2-expressing AT2 cells significantly varied between these two studies likely due to the use of different experimental strategies, but both demonstrated the expansion of this subpopulation after injury and their contribution to alveolar repair [68][69][87,88]. Another subset of AT2 cells is characterized by the expression of CD44 and exhibit high rates of proliferation suggesting a role in alveolar regeneration [67][86]. Whether Axin2+ and CD44+ AT2 cells are the same or a different subpopulation is still to be confirmed.
In addition to AT2 cells, considered as primary contributors to post-injury alveolar regeneration, various epithelial progenitor populations have been shown to participate in regeneration after damage [58][59][70][77,78,89].
BASCs (Bronchoalveolar Stem Cells) are found at the bronchoalveolar duct junction and they have been long understood to be stem cells that give rise to both AT2 cells and Club cells [71][72][90,91]. They were first defined by their anatomical location and the co-expression of the club cell marker, SCGB1A1, and the AT2 cell marker, SFTPC, suggesting their potential to differentiate into both cell types under a condition of lung injury. It was not until recently that it has been demonstrated, using two different approaches to lineage trace these cells in vivo, that indeed BASCs serve as progenitors of the distal airway and alveolar epithelium, being able to generate club cells, ciliated cells, AT2 and AT1 cells, confirming their contribution to lung regeneration [73][74][75][92,93,94].
Besides BASCs, airway epithelial stem/progenitor cell populations have been shown to contribute to both airway and alveolar regeneration [70][71][72][74][76][77][78][79][80][81][82][83][84][85][86][87][88][89,90,91,93,95,96,97,98,99,100,101,102,103,104,105,106,107]. Among these populations, several subsets of club cells in the terminal bronchioles, including Upk3a+ variant club cells, H2-K1 high club cells and MHC-II+ club cells, have been identified to give rise to AT2 cells in different models of severe damage [64][76][78][79][85][83,95,97,98,104].
A rare population of p63-expressing cells was identified in terminal bronchioles and their contribution to airway and alveolar regeneration was examined [80][82][83][86][99,101,102,105]. After severe injury such as bleomycin exposure or influenza infection, this rare population of the airway (Lineage Negative Epithelial Progenitors (LNEPs) [82][101]; Distal Airway Stem Cells (DASCs) [80][99]) activate the expression of Krt5, expand and migrate to damaged regions of the alveoli. However, these cells rarely give rise to AT2 cells nor to AT1 cells [82][86][101,105], but instead form pod-like metaplastic structures in which the alveoli are not repaired which persist for a long time and are likely thought to fill the injured space to preserve structural integrity. Nevertheless, Xi et al. demonstrated using stringent lineage tracing tools that hypoxia and Notch and Wnt signaling regulate the fate decision of p63+ progenitors to either regenerate the alveolar epithelium or to form these pod-like structures [70][89].
Surprisingly, recent evidence demonstrates that even terminally differentiated AT1 cells are able to dedifferentiate and give rise to AT2 cells after pneumonectomy and hyperoxia-induced lung injury [89][90][108,109], reflecting the significant plasticity of lung cells and conferring the lung as a valuable mechanism of repair. A tonic activation of YAP/TAZ is required to maintain AT1 identity, and their loss induces their differentiation into AT2 cells [90][109]. This is of high interest since it reveals mechanisms of the active maintenance of quiescence [48][67] necessary to keep tissue homeostasis similar to the Notch2 tonic activation in Club cells [91][110] that has been previously reported and that is required to maintain their cell identity and preserve adequate proportions of each cell type in the airway epithelium.
2.2. Mechanisms of Alveolar Regeneration
Multiple signaling pathways including BMP, FGF, Notch pathway, Wnt signaling, TGFβ, Hippo pathway, NF-κB and mechanotransduction pathways have been described to regulate AT2 proliferation and/or differentiation [63][66][81][92][93][94][95][96][97][98][99][100][82,85,100,111,112,113,114,115,116,117,118,119]. These signals are provided by the microenvironment, comprised of different cell types including PDGFRα lipofibroblasts, pulmonary endothelial cells and alveolar immune cells, and the extracellular matrix [52][98][101][102][71,117,120,121] which is known to influence stem/progenitor cell behavior in many different tissues including the lung [103][122]. Thus, understanding the communication between AT2 cells and the different cell types in their niche is crucial to unravel the mechanisms involved in lung regeneration during normal physiological repair as well as providing insight into maladaptive repair and pathological processes. Importantly, the precise mechanism regulating the differentiation of AT2 cells into AT1 cells has been recently revealed. Interestingly, the stepwise pathway that an AT2 cell goes through in their way to become an AT1 cell includes the transition through an intermediate state required to achieve successful regeneration. Using a combination of scRNASeq, RNA velocity, lineage tracing, distinct injury models and organoid cultures, three independent groups identified intermediate cells—“pre-alveolar type-1 transitional cell state” (PATS) [104][123], Krt8+ alveolar differentiation intermediate (ADI) cells [88][107] and damage-associated transition progenitors (DATP) [105][124]—characterized by the expression of Cldn4 and Krt8 [88][104][105][107,123,124] in addition to Krt19 and Sfn in PATS [104][123], and these are rarely found in the homeostatic lung but arise after injury. These cells were found in organoids generated by AT2 cells and in a variety of lung injury models including LPS [104][123], bleomycin administration [88][104][105][107,123,124], genetic AT1 cell ablation [104][123], pneumonectomy [104][123], adult hyperoxia and neonatal hypoxia and hyperoxia with Influenza type-A infection [88][107], suggesting that it is a common injury response. Lineage tracing experiments demonstrated that these intermediate cells derive from AT2 cells [88][104][105][107,123,124] with a contribution of airway MHC-II+ club cells that give rise to alveolar lineages through Krt8+ ADI cells [105][124], likely depending on the site and the severity of injury. One of the most striking observations during this process is the activation of pathways associated with stress and this may be the reflect of the significant change in morphology (cell shape and structure) that AT2 cells (cuboidal) need to accomplish in order to differentiate into AT1 cells (flat and thin) [104][123]. Notably, these transitional cells show a unique transcriptional signature characterized by the activation of pathways of DNA damage such as p53 signaling, cellular senescence and TGFβ signaling [66][88][104][105][106][107][108][85,107,123,124,125,126,127]. Interestingly, the pharmacological activation of p53 induced AT2 differentiation, while the genetic inactivation of p53 hampered cells at the transitional stage, demonstrating that p53 signaling is sufficient and necessary to promote AT2 differentiation through PATS to give rise to AT1 cells after injury [104][123]. Importantly, these studies reveal that normal alveolar regeneration involves a transitional state that exhibits features of senescence which usually occurs in aged cells and is thought to be irreversible. Thus, these observations support that reversible physiological senescence may be a program of normal tissue regeneration and maintenance vs. pathological senescence occurring in disease. Previous studies had also observed intermediate states between AT2 and AT1 cells. The accumulation of an AT2 transitional state following pneumonectomy was observed in Cdc42-null mutants, demonstrating that elevated mechanical tension arrested regeneration at an intermediate state and resulted in fibrosis [66][85]. Similarly, the inactivation of Dlk1, a suppressor of Notch signaling, led to the accumulation of an intermediate cell population during AT2 to AT1 cell transition triggered by a Pseudomonas aeruginosa infection, suggesting that Notch activation is needed to initiate alveolar regeneration [100][119]. Although it is unknown whether these cells are similar or distinct from PATS, ADI or DATP cells, altogether these studies confirm that an intermediate AT2-AT1 cell state exists during alveolar regeneration. As previously mentioned, the microenvironment is known to influence AT2 behavior [52][71] and critical cues from their niche drive the initiation of AT2 differentiation. The analysis of the interactome during alveolar regeneration revealed that intermediate cells display a distinct receptor-ligand connectome with mesenchyme and macrophages [88][107]. In fact, this process of AT2 differentiation has been shown to be regulated at least in part by inflammatory cytokines, in particular IL-1β which is produced by interstitial macrophages [105][124], mechanical forces [66][85] and Notch signaling [100][119]. Importantly, the temporal regulation of these signals during AT2-AT1 transition is crucial, since although Notch activation and IL-1β are necessary to initiate AT2 differentiation, their persistence impedes terminal differentiation [100][105][119,124]. Further investigation will reveal other regulatory networks involved in this process, whether other subsets of “regenerative” AT2 cells previously unrecognized such as Il1r1+ AT2 cells [105][124] exist, or how is this differentiation process altered in aged AT2 cells.2.3. Alveolar Regeneration in Human Lungs
Most of ouresearchers' knowledge of lung regeneration derives from studies performed using mouse models. However, important differences exist between the mouse and the human respiratory system. Probably one of the most important differences is the lack of respiratory bronchioles in the mouse lung, which seems to be a key site for the development of diseases. Thus, big efforts are currently being made in order to extend the previous findings into studies of the pathogenesis of human lung diseases such as IPF [88][104][107,123]. Although human alveolar regeneration is poorly characterized, recent studies have identified novel epithelial progenitors with the capacity to regenerate the human alveolar epithelium. Kadur Lakshminarasimha Murthy et al. identified a novel bi-potential cell population that they called AT0 that can normally be found in the alveolar sacs and that emerges and expand after injury [109][128]. Using a non-human primate model of lung injury together with human organoids and tissue samples, they found evidence that during regeneration, AT2 cells transiently go through an AT0 cell state in their differentiation into AT1 cells or secretory cells of the terminal respiratory bronchioles (TRB-SCs). Of note, this process is different than the one observed in the mouse lung and that involves a different transient state known as PATS [104][123]. On the other hand, Basil et al. identified a unique airway secretory cell population in human respiratory bronchioles that they called RAS cells, with the capacity to differentiate into AT2 cells in a process regulated by Notch and Wnt signaling [110][129]. RAS cells are characterized by the expression of SCGB3A2 and lack the expression of the club cell marker, SCGB1A1, and the AT2 cell marker, SFTPC, representing a transcriptionally intermediate state between canonical airway secretory cells and AT2 cells. This novel epithelial progenitor serves as a putative progenitor for the AT2 cell lineage, and the RAS cell-AT2 cell rapid and unidirectional differentiation has been found to be altered in disease such as COPD [110][129]. In chronic lung diseases, mechanisms of repair are dysregulated and inefficient [53][72]. Interestingly, an accumulation of SCGB3A2+ AT2 cells was observed in humans and ferrets that were exposed to cigarette smoke and in COPD lungs [110][129]. Similarly, increased numbers of AT0 cells were found in lungs of patients with COPD and with IPF [109][128]. The accumulation of these transitioning cells could represent a more active attempt to regenerate the damaged tissue, or a blockade in regeneration. The fact that AT2 cells in IPF lungs persistently express markers of intermediate cells [88][104][107,123] suggests that AT2 cells in fibrotic lungs do not have the capacity to further differentiate into AT1 cells [66][107][85,126]. Of note, these intermediate cells also exhibit a profibrogenic signature [88][107], maybe contributing to amplify the fibrotic loop. Importantly, PATS, ADI and DATP signatures were also found in the fibrotic regions of lungs from IPF patients [88][105][109][107,124,128]. Yet, these cells would be more similar to Krt17+ p63+ Krt5- basaloid cells found in sites of active fibrosis, predominantly accumulated at the edge of fibroblast foci in IPF lungs [88][104][111][112][113][114][107,123,130,131,132,133]. These observations suggest that the depletion of these transitioning cells or the promotion of their terminal differentiation into AT1 cells could represent previously unexplored therapeutic avenues. Understanding the tissue-resident stem/progenitor cells involved in lung repair and regeneration post-injury as well as their interactions with the environment and the mechanisms that regulate reparative responses is crucial to developing strategies to promote regeneration and restore functional tissue in chronic lung disease together with therapies that halt the progression of the disease. This knowledge will facilitate the development of new therapeutic approaches for treating respiratory diseases. Strategies for therapeutic lung regeneration are actively pursued and they include: (1) tissue engineering new lungs in vitro [115][134], (2) pharmacological manipulation of endogenous healthy cells to induce repair [48][67], and (3) administration of exogenous stem or progenitor cells to damaged lungs [116][135]. Lung cell transplantation has been achieved in mice following severe injury by influenza infection, by naphthalene injury and followed by systemic irradiation or following a low dose of bleomycin [82][83][117][118][101,102,136,137]. However, these strategies are in the early stages of development and questions about the successful engraftment of these cells, or the actual improvement of lung function remain. In order to apply this therapeutic approach in humans, many open questions need to be answered: What is the best route of cell administration? Is it directly through the airway or by IV injection? Will transplanted cells engraft and survive? How many cells are required? What will be the fate of these cells? Will they differentiate into alveolar epithelial cells in the environment of an injured lung, or will a proper microenvironment also be required to instruct adequate differentiation/regeneration? Which is their mechanism of action? Would these cells exert their effects by engraftment facilitating direct cell-cell contact or through paracrine secretion of growth factors, cytokines and hormones? The transplantation of healthy cells with a regenerative capacity that could contribute to tissue repair is described in further detail below.3. Cell Therapy in Lung Fibrosis
As stated above, pirfenidone and nintedanib are the only drugs approved for the treatment of IPF. Although these two drugs have shown efficacy in reducing the rate of decline in lung function and slowing the pace of the disease’s progression, they are not able to halt the disease’s progression [119][120][138,139]. In addition, both compounds have shown significant side effects such as gastrointestinal- and skin-related adverse events in the case of pirfenidone, and diarrhea in the case of nintedanib [119][120][138,139]. In order to cure pulmonary fibrosis, rwesearchers must probably combine therapies addressed towards blocking pathogenesis and promoting tissue regeneration to completely restore lung tissue that is able to successfully function. Stem cells and endogenous lung progenitor cells have been studied for many years as a therapy to promote tissue repair in chronic diseases because of their regenerative potential including their self-renewal capacity and ability to differentiate into different cell types of their tissue [121][140]. Remarkably, stem cell-based tissue engineering aims to mimic the native stem cell niche and maintain stem cell function within the graft by providing appropriate microenvironmental cues in a controlled and reproducible fashion to facilitate its application into human diseases including IPF [122][141]. In the last decade, cell therapy has been investigated for the treatment of IPF, including the use of a variety of cell types such as lung epithelial cells, specifically AT2 cells [123][142], induced pluripotent stem cells (iPSCs), and mesenchymal stem cells (MSCs) isolated from bone marrow stroma [124][143] and those from adipose tissue or from other tissues (Figure 3; Table 2). Notably, both endogenous alveolar epithelial and mesenchymal stem cells are the most widely investigated ones for the treatment of IPF.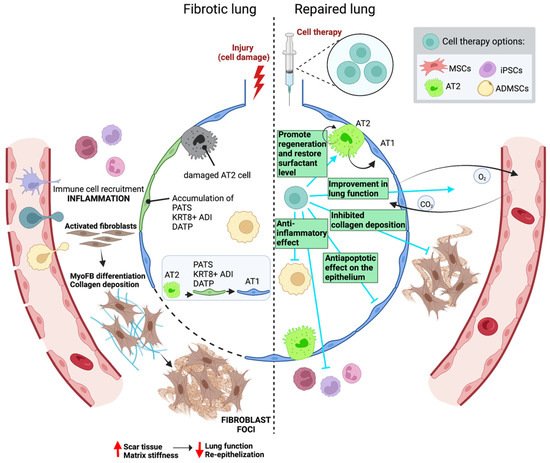
3.1. Preclinical Mouse Studies
3.1.1. Epithelial Cells: Alveolar Type 2 Cells
AT2 cells function as stem cells in the adult lung [59][78], maintaining tissue homeostasis and contributing to alveolar repair after damage [125][144]. However, in IPF, a significant portion of AT2 cells are lost or are dysfunctional and are replaced by fibroblasts and myofibroblasts [123][142]. In previous preclinical studies, Serrano-Mollar and colleagues revealed the therapeutic effect of the intratracheal transplantation of AT2 cells resulting in a reduction of the extent of experimental pulmonary fibrosis [126][127][145,146]. AT2 cell treatment at either 3, 7 or 15 post-bleomycin installation showed decreased fibroblast proliferation and prevented accumulation of the ECM [127][146]. Furthermore, the injection of AT2 cells 14 days post bleomycin-induced lung injury resulted in a restoration of the surfactant levels [126][145] and similarly, the administration of AT2 cells 7 days after injury improved the lung function based on elastance and compliance measurements [128][147]. Interestingly, Cores and colleagues established that adult lung spheroid cells (LSCs) are an intrinsic source of therapeutic lung stem cells [129][148]. The administration of these cells right after bleomycin instillation in a rat model decreased the extent of fibrosis, reduced apoptosis, protected alveolar structures and increased angiogenesis [129][148].3.1.2. Adult Mesenchymal Stromal/Stem Cells
Adult mesenchymal stromal/stem cells (MSCs) are multipotent cells with the ability to differentiate into a wide range of cell types [130][131][149,150]. Not only murine, but also human MSCs have been used in studies involving animal models [132][133][151,152]. According to several studies, MSCs can home to damaged tissues when systemically administered via intravenous (IV) or intraperitoneal (IP) injection [134][135][153,154]. Ortiz and colleagues reported for the first time that bone marrow MSC-derived cells (BM-MSCs) injected into the jugular vein immediately after bleomycin instillation were able to engraft to sites of lung injury, reducing both inflammation and collagen deposition [136][155]. In contrast, beneficial effects were not observed when they were given 7 days after the injury occurred [136][155]. Similarly, other studies also showed that only the very early administration of BM-MSC after bleomycin decreased the damage that incurred [137][138][139][140][141][156,157,158,159,160]. Moreover, the combination of BM-MSC with nintedanib in bleomycin-induced lung fibrosis in rats showed anti-inflammatory and antifibrotic activity [142][161]. Furthermore, tissue-resident MSCs have also been shown to protect lung integrity after bleomycin instillation in mice [143][162]. MSCs have been extensively used in a number of clinical trials to find treatment options for many different diseases. In fact, it has been shown that MSCs have potent anti-proliferative, anti-apoptotic, immune-modulatory and anti-inflammatory properties, besides their multilineage differentiation capacity, making them an attractive and promising option for diseases with no cure [130][144][149,163]. However, the majority of the preclinical studies administering BM-MSCs have used them as a preventive strategy rather than a treatment approach, and further research analyzing their effects after injury are required in order to validate their therapeutic potential [136][145][146][155,164,165]. The major limitation to evaluating the potential of BM-MSCs as a treatment for lung fibrosis is that these studies use the single-dose bleomycin model which induces reversible damage and fibrosis resolves naturally, narrowing the time window for these cells to exert their action. Thus, a model of established fibrosis like the chronic administration of bleomycin to resemble some of the major pathological features of human IPF [45][46][45,46] must be used to test their efficacy as a treatment. Yet, it is important to take into account that most of the studies were performed in rodents subjected to established injury models (i.e., bleomycin administration) that usually display a severe phenotype but do not reflect genetic or inter-strain variability and do not completely mimic the pathophysiology of the human disease [147][148][166,167]. Adipose mesenchymal stem cells (ADMSC) have been also evaluated [149][150][151][152][153][168,169,170,171,172]. The administration of adipose MSCs 7 days post-bleomycin treatment inhibited both a pulmonary inflammation and fibrosis which significantly improved the survival rate of mice in a dose-dependent manner [150][169]. In concordance, Lee and colleagues observed a reduction in the hyperplasia of epithelial cells and a reduction in the inflammation and fibrosis after the repeated ADMSC administration at 8, 10, 12 and 14 weeks in a chronic model of fibrosis [151][170]. Hence, ADMSCs could have positive effects even when they are administered once fibrosis has been established. A study using human derived ADMSC demonstrated that these cells significantly increase survivability and reduce organ weight and collagen deposition better than pirfenidone does after the intratracheal challenge with bleomycin in mice [153][172]. In general, ADMSCs have proven their efficacy in bleomycin-treated aged mice when they come from a young donor [152][171]. Interestingly, the age-dependent antifibrotic properties of these cells was demonstrated since old donor-ADMSC treatment in old bleomycin-treated mice did not reduce fibrosis and related markers [150][169]. Furthermore, another study used human placental mesenchymal stem cells of fetal origins (hfPMSCs) and this showed that inflammation and fibrosis in bleomycin-treated mice was alleviated after treatment with these cells [154][173]. Similarly, amniotic fluid stem cells (AFSC) have shown the potential to inhibit the development or progression of the fibrotic phenotype in both acute and chronic remodeling events in mice treated with bleomycin [155][174].3.1.3. Induced Pluripotent Stem Cells
Several studies have transplanted pluripotent stem cells to bleomycin-treated animals to evaluate their antifibrotic and/or alveolar regenerative efficacy [156][157][158][159][160][161][162][163][175,176,177,178,179,180,181,182]. Previous studies have demonstrated that the administration of pluripotent- induced stem cells (iPSCs) to mice 24 h after a bleomycin challenge reduced the development of lung fibrosis [156][157][175,176]. The optimization of methods to successfully differentiate human embryonic stem cells (hESCs) [161][180] and pluripotent stem cells [158][159][162][177,178,181] into alveolar epithelial cells has allowed for the use of these derived cells for cell-based approaches. A mixture of epithelial cells (AT2, AT1 and Club cells) derived from hESCs and administered 7 days after bleomycin instillation showed a reversion in the fibrotic phenotype [161][180]. The authors concluded that engrafted cells may reduce fibrosis either by directly replacing the fibrotic tissue or indirectly by paracrine secretion of antifibrotic factors [161][180]. Improved approaches include the use of AT2 cells derived from iPSCs-induced pluripotent stem cells (iPSCs). The administration of mouse iPSC-derived AT2 cells 24 h after bleomycin instillation reduced the extent of the fibrosis since the collagen content was decreased and the lung tissue structure recovered [160][179]. Importantly, differentiated iPSCs labeled with a PKH26 cell tracker that were engrafted into the lungs and gave rise to AT2 and AT1 cells, recovering the cell numbers observed in control mice. Furthermore, inflammatory cell infiltration, as well as TNFα and IL-6 were reduced [160][179]. Interestingly, the transplantation of human iPSC-AT2 cells 15 days after bleomycin instillation, when fibrosis was already present in the lungs, induced a reduction in the collagen deposition by the inhibition of both TGF-β and α-SMA expression, and no evidence of inflammation or epithelial damage was observed in the transplanted lungs [163][182]. Consistently, fewer αSMA+ myofibroblasts developed, and fewer and smaller fibroblast foci were observed [163][182]. Of note, engraftment of these cells was not detected, so a paracrine effect is suggested to explain this. These findings reveal a potential use of iPSC-AT2 cells to resolve fibrotic damage. However, although this enstrudy demonstrates that the administration of iPSC-AT2 cells at the fibrotic stage of bleomycin-induced injury halts and reverses fibrosis, a more appropriate model of established fibrosis is necessary to test the actual potential of these cells to be used as a therapy for the treatment of IPF. The use of AT2 cells derived from iPSCs is, undoubtedly, an easy and efficient approach for the lung cell-based therapy that allows for autologous cell transplantation. However, although cells derived from iPSCs bypass the ethical concern associated with the use of human embryonic stem cells, it is necessary to establish protocols to ensure their correct administration and engraftment to guarantee their beneficial effects and to prevent dangerous side effects including the formation of tumors [164][183].3.2. Clinical Human Studies
3.2. Clinical Human Studies
Epithelial and mesenchymal stem cells have been used in different clinical trials. The administration of human AT2 cells to IPF patients was demonstrated to be safe, well tolerated and showed no relevant side effects in patients with moderate and progressive IPF, during the supervised 12-month period [142]. However, it has not been assessed yet, whether this treatment provides an improvement in lung structure and functionality [142].
Interestingly, basal cells (BCs) have also been tested as a potential cell-based strategy for lung diseases. BCs act as airway stem cells that can give rise to all the epithelial cell types present in the mouse trachea and human airway epithelium [184,185]. Of note, they have shown to be able to migrate to lower parts of the respiratory tree and contribute to alveolar regeneration in cases of severe injury [101,106]. In this regard, a rare subset of human airway BCs characterized by the expression of SOX9 and with the capacity to regenerate functional alveoli has been identified [186,187]. Interestingly, the transplantation of autologous adult human SOX9+ airway BCs into bronchiectasis patients improved their lung function and no aberrant cell growth nor other related adverse events were observed during the whole follow-up period [186,187]. Hence, the use of airway basal cells in stem cell therapy for lung diseases may provide new opportunities to regenerate damaged lung tissue. Despite this strategy having not being tried for IPF, p63+ progenitors may represent an additional source that may give rise to alveolar lineages. Yet, to be used therapeutically, hypoxia, Notch and Wnt signaling should be regulated to avoid these cells from forming metaplastic structures with Krt5+ cells [65,77].
MSCs have been more extensively used. In human IPF, Averyanov and colleagues evaluated the safety, tolerability and efficiency of high cumulative doses of MSCs in fibrotic lungs and observed a rapid progressive course of severe to moderate IPF without significant adverse effects or differences in mortality after the administration of MSCs [188]. Another phase one trial study confirmed the safety of MSC (specifically autologous adipose-derived stromal cells (ADSCs)-stromal vascular fraction (SVF)) application, and reported improvements in life quality parameters and promising progression-free survival rates up to 24 months in 14 IPF patients, concluding that further and more complex clinical trials are needed to decipher the role of ADSCs in IPF pathogenesis and treatment [189,190]. Further phase one trials in nine patients with mild or moderate IPF and eight patients with moderately severe IPF obtained similar results [191,192]. Although a follow-up period of six months did not show improvements in lung function parameters and CT scores [191], the follow up 48 weeks post-transplantation revealed hints of therapeutic improvements with slower progression of fibrosis scores measured by CT scans and a slower decrease in the lung diffusion capacity for carbon monoxide in those patients receiving the higher MSC dosage [193]. Results about the potential of MSC administration as a preventive or curative therapy are heterogeneous, and in some cases contradictory due to differences in the specific cell type, dosage and the route of administration [194,195,196]. Future research efforts need to be addressed towards establishing the optimal conditions and defining the therapeutic window wherein treatment with these cells may be beneficial.
Table 2. Cell therapy in preclinical mouse studies and clinical human studies.
4. Current Challenges
Idiopathic pulmonary fibrosis is a rare and fatal disease with no cure. Unfortunately, there are still a lot of unanswered questions about its pathophysiology and the mechanisms underlying its initiation and progression, and little progress would be possible without proper models to study IPF. In vivo models rely on measuring the response to injury caused by bleomycin, a tumor chemotherapeutic agent that has been used to study pathological fibrosis in the lung for around 50 years [197]. The lack of treatment options and the heterogeneity in the natural progression of the disease among patients highlights the importance of developing new strategies, not only to tackle IPF emergence and progression, but also to try to reverse fibrosis and restore functional tissues. The ideal therapeutic strategy should first, eliminate the source of the injury, although in most cases this is unknown. Next, it should remove the fibrotic tissue that is precluding an efficient gas exchange and physiological cellular crosstalk within alveoli; and third, it should promote the regeneration of damaged tissues to restore tissue function and recover a homeostatic state. In this regard, cell therapy offers great opportunities towards disease healing. Nonetheless, we must take into account that most of the cell replacement therapy strategies have been assayed as a pretreatment in preclinical studies, being administered in the inflammatory stage (24 h-7 days after bleomycin challenge) rather than in the fibrotic phase which impedes an assessment of the efficacy of the transplantation of the different cell types in resolving fibrosis [198,199]. In general, cell transplantation during these initial stages showed promising results since the progenitor cells that were administered inhibited inflammation and impeded fibrogenesis. However, controversial results are found when cells were administered during the fibrotic phase, raising questions about their actual therapeutic utility [198,199]. These issues most likely arose due to the injury model used, since the single-dose bleomycin model is an acute injury model that resolves naturally and therefore, cannot be compared to the chronic and progressive nature of human IPF. As aforementioned, a better model of established fibrosis needs to be used to analyze the therapeutic potential of transplanted cells in halting and/or reversing fibrosis.
All in all, there are still limitations in the use of stem cells for therapy, for example, the ability of exogenous stem cells to home into the desired tissue has been shown to be relatively low. Currently, the best strategy to promote tissue regeneration seems to be to promote endogenous cells to regenerate lost or damaged tissue. However, there is still much work ahead to unravel how these cells achieve regeneration successfully and which cells do it best.
5. Future Perspectives and Conclusions
The remarkable plasticity that airway and alveolar epithelial cells exhibit provides the lung with a wide variety of region-specific stem and progenitor cell populations that participate in regeneration after injury. Leveraging the mechanisms governing lung regeneration may help to develop strategies aimed towards inducing tissue repair in the diseased lung.
Of note, just as important as identifying the reparative cells that can contribute to the process of lung regeneration is the understanding of the microenvironment that dictates the path of an endogenous or exogenous progenitor. Understanding the interactions between epithelial progenitors and their niche and how these signaling inputs are integrated is crucial to enhance inefficient mechanisms of repair in areas of advanced fibrosis where scar tissue impedes the survival of healthy cells.
Over many years, replacing pathologic epithelial cells with exogenous progenitors has been thought to be a promising treatment for fibrotic pulmonary diseases. However, the potential of stem cell-based therapy is still not a reality despite significant advances accomplished in the field. Currently, a better knowledge about which cells can execute regenerative responses as well as the development of optimized methods for expanding lung progenitors encourage the clinical application of these therapies. Yet, more mechanistic research is needed in order to demonstrate the actual potential of epithelial progenitor transplantation in patients with lung fibrosis as well as the long-term fate of these cells, including the possibility of their contribution to the development of tumors.
Research towards the study of the mechanisms of quiescence to return to steady-state conditions with the active signals and interactions that maintain tissue homeostasis is of paramount importance in order to restore tissue balance after the therapeutic induction of lung regeneration when tissue repair has been achieved.
In addition to the administration of exogenous cells, the stimulation of endogenous progenitors has also been proposed as a therapeutic approach to promote lung regeneration. Lessons from lung development and postnatal alveologenesis may provide clues to developing this strategy. However, other limitations and unanswered questions for the successful achievement of the therapeutic induction of lung regeneration exist. First, it is unknown whether endogenous AT2 cells in fibrotic lungs are able to accomplish successful lung regeneration. Many questions arise because of this; how damaged are they? Do they retain the capacity to proliferate and initiate differentiation into functional AT1 cells? Are they going to find the way to initiate self-organization of functional alveoli? If this process is instructed by ECM that is highly altered in IPF, is it going to be possible to regenerate functional alveolar units with an adequate cell distribution in this context? Should the microenvironment be restored first, so signals received by progenitors are the appropriate ones necessary for regeneration?
A better understanding of human lung regeneration is required. The development of new models is particularly necessary to investigate the mechanisms involved in repair and regeneration and to find out how to promote lung regeneration to treat human lung disease. Larger animal models, such as those involving non-human primates, ferrets and pigs may be necessary for this purpose as their respiratory anatomy closely resembles the anatomy of the human respiratory system. Alternatively, precision cut lung slides (PCLS) from healthy and IPF lungs allow for the study of human lung progenitors and regeneration in the human lungs. These models promise to close the gap between basic research and clinical application, providing the basis for a deeper understanding of lung physiology and pathology in humans and allowing the design of more curative therapies.
Much research is still needed in order to develop successful strategies focused on cell-based therapies to promote lung regeneration in combination with other therapeutic approaches, targeting pathological events and addressed towards eliminating aberrant cells and the matrix to restore lung architecture and regenerate functional alveolar units for breathing.