Cystic fibrosis (CF) is still today the most common lethal genetic disease with autosomal recessive inheritance in the Caucasian population, with a prevalence of 1 case per 2500 live births. The disease is caused by a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that causes the CFTR protein to become dysfunctional. When the protein is not working correctly, there is reduced transport of chloride ions with consequent dysregulation of epithelial lining fluid (mucus) transport in the lung, pancreas and other organs. Oxidative stress is a complex process in which excess reactive oxygen species (ROS) affect, either directly or indirectly, all structural and functional components of cells at a molecular level. This arises because the production of these chemical species is increased and/or because the physiological defense capacity towards them, thanks to the antioxidant system, is reduced.
1. Evidence from Literature
One of the main determinants of progressive lung damage in CF is represented by chronic oxidative stress, which leads to the establishment of an intrinsically proinflammatory environment. The mechanisms behind this are still partially unknown, but several studies have shown the direct implication of CFTR protein dysfunction, mainly in the lungs, but also in extrapulmonary tissues such as the pancreas and intestine
[1].
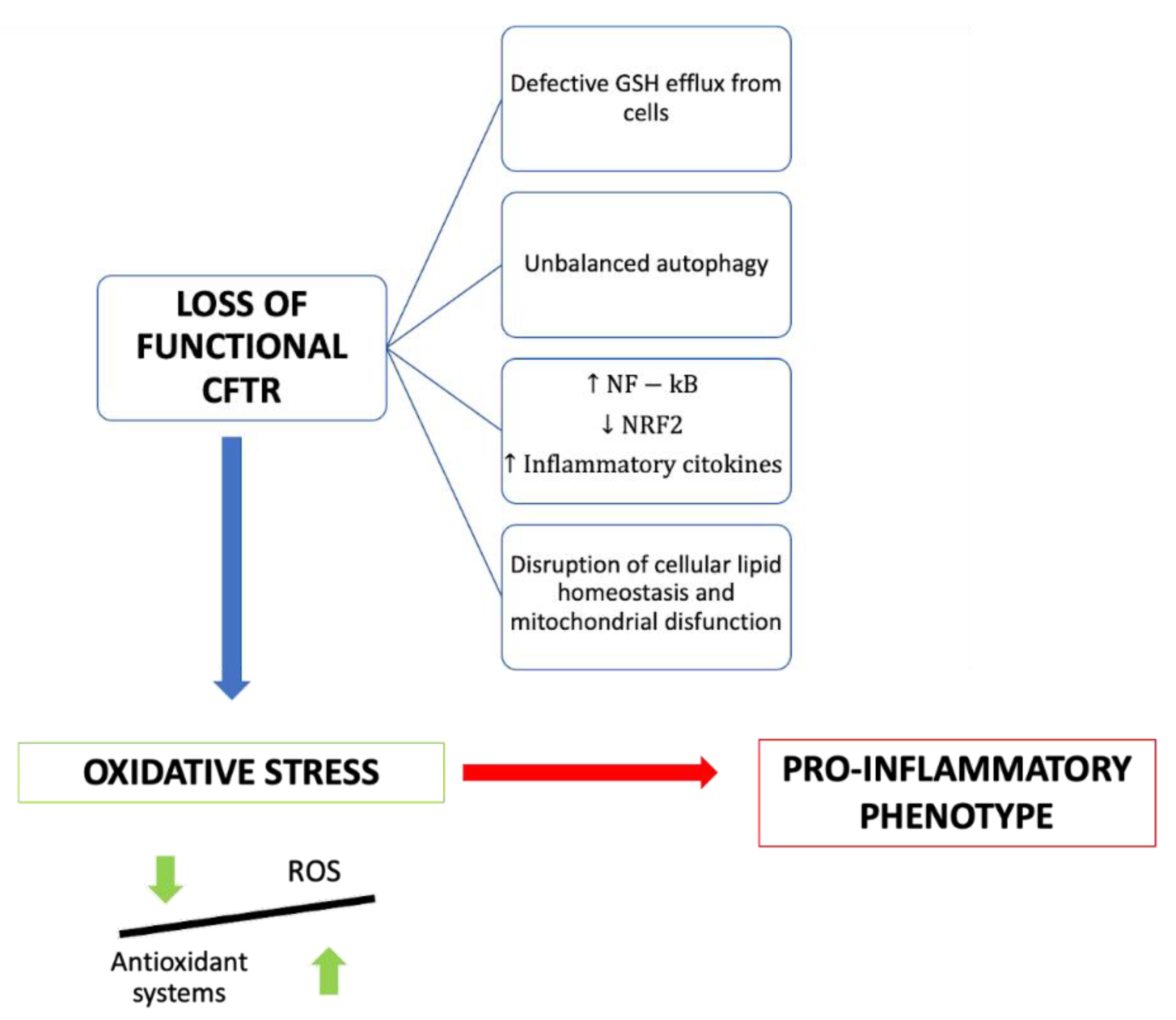
Several molecular mechanisms have been proposed to explain the link between CFTR deficiency and oxidative stress (Figure 1).
Figure 1. Summary of the consequences of the loss of functional CFTR in cystic fibrosis patients.
There is some evidence that the efflux of GSH out of cells is a chloride-dependent mechanism involving the CFTR channel. Indeed, CFTR shares a structural similarity with ABCC proteins, which normally export glutathione and/or glutathione S-conjugates
[2]. Glutathione (GSH) is a tripeptide with antioxidant properties consisting of cysteine, glycine and glutamic acid. It represents the first-line defense of the lung against oxidative stress-induced damage, and its availability inside the cell is fundamental to sustaining a good redox state. The ratio between reduced and oxidized glutathione is an indicator of the cellular redox state and describes the antioxidative capacity of cells
[3].
Unsurprisingly, in patients with CF, low CFTR activity is correlated with GSH deficiency, resulting in an altered extracellular ratio between oxidized and reduced glutathione
[4][5][6]; oxidized glutathione species are significantly elevated, and there is an inadequate response to neutrophil-mediated oxidative stress during infections. Rather, the reactive oxidant species produced by neutrophils, including myeloperoxidase (MPO)-derived hypochlorous acid, contribute to the oxidation of glutathione, leading to a vicious cycle. Dickerhof N et al. demonstrated that the pharmacological inhibition of MPO by orally administrated AZM198 decreases oxidative stress and improves infection outcomes in mice with CF-like inflammation without interfering with the clearance of bacteria
[7]. Still, few studies have been conducted or are ongoing on the beneficial effects of direct GSH supplementation in CF. For example, Calabrese et al. studied the possible beneficial effects of long-term treatment with inhaled glutathione
[8]. Hewson et al. established that exogenous administration of γ-glutamylcysteine (GGC), the immediate precursor of glutathione, can increase intracellular levels of total glutathione and protect CF cells from lipopolysaccharide (LPS)-induced cell damage
[9].
It has also been demonstrated that the administration of N-acetylcysteine (NAC), the acetylated form of the amino acid L-cysteine and a precursor to glutathione, is able to reduce the redox imbalance, increasing the GSH level. Furthermore, an influence of NAC on nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) activation was observed
[10].
Mutated CFTR is associated with the alteration of some signal transduction pathways at a cellular level, such as that of NFkB, required for the transcription of various proinflammatory molecules. NFkB overexpression is an intrinsic underlying feature of the patient with cystic fibrosis and is exacerbated by hyperproduction of ROS and by bacterial stimulation on the cell surface that induces further activation. Moreover, the CFTR mutation is also associated with reduced production of peroxisome proliferator-activated receptor (PPAR), a transcription factor that normally counteracts the action of NFKB
[1][10]. This results in increased production of oxidizing molecules and proinflammatory cytokines such as IL-1, TNF, IL-6 and IL-17A.
In normal cell physiology, under conditions of increased oxidant production, a series of pathways are activated that play an active role in the suppression of inflammatory signaling. Among these, the most important is the Nuclear factor erythroid 2-related factor 2 (Nrf2) pathway, an antagonist of proinflammatory transcription factors such as NFkB. Following the hyperactivation of the inflammatory response, the Kelch-like-ECH-associated protein 1 (KEAP1) protein, which normally binds Nrf2 in the cytoplasm of cells, oxidizes and dissociates from Nrf2, allowing its subsequent transcriptional activation, which leads to the production of over 200 antioxidant and detoxifying proteins; these include heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase 1 (NQO1), glutamate–cysteine ligase (GCL) and glutathione S transferase (GST). Nrf2-mediated HO-1 expression is also regulated by transcription factor BTB (TF BTB) and CNC Homology 1 (Bach1), which suppress HO-1 expression
[8][11]. The heme oxygenase-1/carbon monoxide (HO-1/CO) pathway is essential to ensure a controlled immune response and effective bactericidal activity by monocytes and macrophages. The blunt activation of this pathway in CF patients therefore contributes to hyperinflammation and defective host defense against bacteria. Recent studies have shown that the administration of controlled doses of CO can induce HO1 by reducing lung hyperinflammation and oxidative stress
[12]. Furthermore, CO stimulates autophagy
[12], the cellular mechanism that is fundamental to efficient bacterial clearance by immune cells. Recent works prove that CFTR deficiency in macrophages and neutrophils results in an inability to kill bacteria and, thus, in limited autophagy activity
[13].
There is much evidence that the Nrf2 pathway is dysfunctional in cells with mutated CFTR
[2][11][12].
Laselva et al. demonstrated that the administration of dimethyl fumarate (DMF), an activator of the Nrf2 pathway, drastically reduced both the basal and stimulated expression of proinflammatory cytokines while also exerting an antioxidant effect
[13].
Borcherding et al. demonstrated that the CFTR modulators VX-809 (Lumacaftor) and VX-661 (Tezacaftor) significantly increase Nrf2 activity in CF patients
[14]; this could represent one of the mechanisms through which CFTR modulators mitigate the inflammatory response and oxidative stress.
Pellullo et al., in their study, used mRNA extracted from nasal epithelial cells to analyze the expression levels of the genes involved in oxidative stress. They found that the expression of Nrf2 mRNA and its targets, such as HO-1 and miR-125b, is upregulated in the nasal epithelia cells of CF patients compared to healthy subjects. This suggests that the protective mechanisms against oxidative stress may be functional but not sufficient to counteract the hyperproduction of ROS and the oxidative stress that characterizes the pathology. Moreover, the authors found that elevated HO-1 and miR-125b levels are associated with an improved FEV1 value, so they could be considered potential predictive biomarkers of CF clinical outcomes. The wide expression range of these markers could partly explain the phenotypic variability of CF, beyond the mutations of the CFTR gene itself
[11].
Another mechanism that links CFTR deficiency and oxidative stress and that contributes to CF airways’ chronic damage is the alteration of lipid metabolism
[12]. Thanks to several studies, it has been found that CF airway pathology is related to alterations in fatty acids, ceramides and cholesterol, but their role in the etiopathogenesis of CF pulmonary pathology is unclear.
An increased ratio of long-chain to very long chain ceramide species (LCC/VLCC), abnormalities in sphingosine phosphate (S1P) metabolism and, consequently, abnormal lipid levels in the blood and lungs are hallmarks of CF. Lipid synthesis is increased, whereas their catabolism is reduced, contributing to inflammation, oxidative stress and impaired autophagy. In this view, dyslipidemia should be considered a contributor to CF airways’ chronic damage, and thus, lipid metabolism should become an important therapeutic target
[15][16][17].
Signorelli et al. have shown that modulating the synthesis of sphingolipids and hindering the accumulation of ceramide with Myrocin (Myr), a sphingolipid synthesis inhibitor, significantly reduces the accumulation of lipids, promotes the oxidation of fatty acids and reduces inflammation and oxidative stress, and it restores the defensive response against pathogen infection, which is defective in CF
[18].
Veltman et al. examined the consequences of CFTR deficiency on lipid metabolism, highlighting how the alterations concerning the metabolism of fatty acids and ceramide induce a state of chronic oxidative stress, but also, in turn, chronic oxidative stress can cause a great imbalance in the metabolism of lipids. The new CFTR-modulating drugs considerably reduce the alterations of lipid balance, confirming the role of CFTR as a regulator of cellular lipid balance
[19].
2. InsOxidatightful Analysive Stress in CF Patients
The role of oxidative stress in the progression of lung injury in CF patients has been widely recognized and very well described in the literature. It has been shown that in patients with CF, there is an important deficit of antioxidant molecules and an increase in oxidative stress
[6][20]. The sustained imbalance between oxidant and antioxidant species induces chronic inflammation, which is the key element contributing to persistent cellular damage and preventing proper airway remodeling. Both hereditary and acquired factors, such as CFTR deficiency and persistent infections, contribute to abnormal and self-sustaining lung inflammation in CF.
The central role of CFTR deficiency has been increasingly recognized in recent years. Mutations in the CFTR gene appear to make CF epithelial cells more susceptible to inflammation compared with healthy cells; for this reason, once an infection is introduced, it triggers the onset of mucosal damage and chronic airway infection. CFTR dysfunction, in fact, not only alters ion exchange and fluid secretion in the lungs but also causes the dysregulation of several signaling pathways, generating an innate oxidative state that, over time, could promote the loss of lung epithelial cell integrity. Impaired extracellular glutathione transport, alterations in lipid metabolism, dysregulation of the main pro- and anti-inflammatory signaling pathways and unbalanced autophagy are the main molecular mechanisms correlated with CFTR dysfunction in CF. Starting from this consolidated knowledge, numerous research groups have identified targets and strategies aimed at reducing the exaggerated immune response that causes chronic inflammation in CF, without altering the natural defenses against infection. Currently used drugs, such as steroidal and non-steroidal anti-inflammatories, mucolytics and antibiotics, reduce inflammation, improving the natural history of the disease; however, there are a lot of concerns about their chronic use because of their immunosuppressive effects that compromise the host’s defenses
[12].
New therapeutic approaches are therefore needed and are currently being evaluated, with the aim of reducing the proinflammatory response in CF, preserving the host defense against microorganisms. The most promising results come from the use of CFTR modulators, which, in the last several years, have radically changed the natural history of CF. The researchers here found that the excellent results from the use of these drugs are related not only to the restoration of physiological ion exchanges, which improve mucociliary clearance, but also, given the close correlation between CFTR deficiency and oxidative stress, to the reduction in the basic inflammatory status that characterizes CF patients. Further studies are needed to confirm, through the evaluation of specific markers, whether CF patients treated with modulators have a significant reduction in oxidative stress. However, these therapies are targeted for specific mutations characterizing CF, and it is not known how long they will have this crucial role in containing the inflammatory response. Future therapeutic perspectives should include the use of additional antioxidant and anti-inflammatory drugs, in combination with CFTR modulators, which specifically target the altered signaling pathway, in order to obtain a more selective response without altering the local tissue defenses.