Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Harald Wajant and Version 2 by Beatrix Zheng.
Tumor necrosis factor (TNF) receptor associated factor-2 (TRAF2) is an intracellular adapter protein with E3 ligase activity, which interacts with a plethora of other signaling proteins, including plasma membrane receptors, kinases, phosphatases, other E3 ligases, and deubiquitinases. TRAF2 is involved in various cancer-relevant cellular processes, such as the activation of transcription factors of the NFκB family, stimulation of mitogen-activated protein (MAP) kinase cascades, endoplasmic reticulum (ER) stress signaling, autophagy, and the control of cell death programs. In a context-dependent manner, TRAF2 promotes tumor development but it can also act as a tumor suppressor.
- apoptosis
- autophagy
- B-cell lymphoma
- cellular inhibitor of apoptosis 1/2 (cIAP1/2)
- necroptosis
1. Introduction
In pioneering work in the mid-1990s, the group of D. Goeddel identified four proteins recruiting to tumor necrosis factor (TNF) receptor 2 (TNFR2). Two of these proteins indicated homology to the just previously identified baculovirus-encoded inhibitor of apoptosis proteins and were accordingly named cellular inhibitor of apoptosis 1 (cIAP1) and -2 (cIAP2) [1]. The two other proteins demonstrated no homologies to proteins known at that time, but shared a conserved C-terminal stretch of app. 200 aa. The two proteins have been named TNF receptor-associated factor-1 (TRAF1) and -2 (TRAF2) and the C-terminal homology domain accordingly as TRAF domain [2]. A C-terminal TRAF domain has also been discovered in four other human proteins, named TRAF3 to TRAF6, and has been subdivided in the compact coiled-coil TRAF-N domain mediating trimerization, and the more loosely packed TRAF-C domain, which in the case of TRAF2 mediates binding to a short aa motif in the cytoplasmic domain of TNFR2 (Figure 1). With exception of TRAF1, the TRAF proteins also share a common N-terminal domain architecture composed of an interesting new gene (RING) domain followed by 5-7 zinc fingers. While activated TNFR2 directly binds TRAF2 and TRAF1, cIAP1 and cIAP2 are indirectly recruited to TNFR2 by help of TRAF2. In fact, eventually TRAF2 seems to fulfill many of its functions in concert with these proteins.
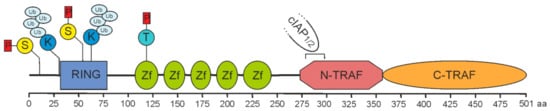
Figure 1. Domain architecture of TRAF2. A sequence of 501 amino acids prescribes the molecular structure of full-length TRAF2. It is essentially composed of a RING domain (aa 32–79), five zinc fingers (Zf) and a TRAF domain comprising a coiled-coil N-TRAF domain with a cIAP1/2 binding site (aa 283–294) [3][4][3,4] and a C-TRAF domain. Phosphorylation and ubiquitination sites of known relevance for TRAF2 function are indicated and comprise serine S11 and S55 [5][6][7][5,6,7], lysine K31 and K63 [8][9][8,9], and threonine T117 [10].
Early after its identification as part of the TNFR2 signaling complex, it has been recognized that TRAF2 is also recruited to the majority of other receptors of the TNF receptor superfamily (TNFRSF) including nearly all TNFRSF receptors (TNFRs) with a TRAF binding motif (direct TRAF2 binding, see Table 1) and all TNFRs with a death domain (DD) (indirect TRAF2 binding). Later, it also became evident that TRAF2 acts beyond the TNFRSF in the signal transduction by other immune regulatory receptors, including members of the toll-like receptor (TLR) family, the type I interferon receptor and the retinoic acid-inducible gene I (RIG I)-like receptor (RLR) family of intracellular pattern recognition receptors recognizing viral RNA (Table 1). Finally, yet importantly, TRAF2 has been implicated in autophagy and endoplasmic reticulum (ER) stress signaling. TRAF2 fulfills its functions primarily by acting as a scaffold, which in a signal-inducible or constitutive manner brings together E3 ligases, their substrates, and also a variety of regulatory factors, including deubiquitinating enzymes (Table 1).
Table 1. TRAF2 interacting proteins.
| Protein | Type of Protein | Experimental Evidence |
Target Domain in TRAF | Reference | |
|---|---|---|---|---|---|
| TNFR2 | TNFRSF | THS, GST, endo Co-IP | CTD | [2] | |
| LTβR | TNFRSF | endo Co-IP | [11] | ||
| OX40 | TNFRSF | THS, Co-IP | [12][13] | [12,13] | |
| CD40 | TNFRSF | THS, GST, endo Co-IP | [14] | ||
| CD27 | TNFRSF | THS, Co-IP | [15][16][17] | [15,16,17] | |
| CD30 | TNFRSF | THS, GST | [18][19] | [18,19] | |
| 4-1BB | TNFRSF | THS, GST, endo Co-IP | [12][20][21] | [12,20,21] | |
| RANK | TNFRSF | GST, Co-IP | [22][23][24] | [22,23,24] | |
| Fn14 | TNFRSF | GST | [25] | ||
| TACI | TNFRSF | THS, Co-IP | [26] | ||
| HVEM | TNFRSF | GST | [27] | ||
| NGFR | TNFRSF | Co-IP | [28] | ||
| BCMA | TNFRSF | Co-IP | [29] | ||
| GITR | TNFRSF | THS, endo Co-IP | [30][31] | [30,31] | |
| TROY | TNFRSF | Co-IP | [32] | ||
| IL15R | receptor | Co-IP | [33] | ||
| IFNαR1 | receptor | GST, Co-IP | [34] | ||
| LMP1 | viral oncogen | GST, Co-IP | [35][36] | [35,36] | |
| A20 | DUB, E3 ligase | THS, Co-IP | [37] | ||
| AIP1 | Ras-GAP | Co-IP | RING/zinc | [38] | |
| AIMP2 | adaptor | THS, GST, endo Co-IP | [39] | ||
| APPL1 | adaptor | GST, endo Co-IP | [40] | ||
| AWP1 | adaptor | THS, Co-IP | TD | [41] | |
| Bcl10 | adaptor | THS, Co-IP | [42] | ||
| Beclin | autophagy | GST, endo Co-IP | RING | [43] | |
| Caspase-2 | caspase | Endo Co-IP | [44] | ||
| Caspase-12 | caspase | Co-IP | NTD | [45] | |
| β-catenin | proto-oncogene | Co-IP, MST | TD | [46] | |
| caveolin-1 | plasma membrane protein | endo Co-IP | [47] | ||
| CHIP | E3 ligase | endo Co-IP | [48] | ||
| cIAP1 | E3 ligase | THS, Co-IP | NTD | [1] | |
| cIAP2 | E3 ligase | THS, Co-IP | NTD | [1] | |
| CYLD | DUB | THS, Co-IP | TD | [49] | |
| DUSP14 | phosphatase | Co-IP | [50] | ||
| DYRK1A | kinase | endo Co-IP | TD | [51] | |
| EGFR | kinase | endo Co-IP | [52] | ||
| EI24 | E3 ligase | Co-IP | [53] | ||
| eIF4GI | scaffold | THS, GST, Co-IP | TD | [54] | |
| Eva1 | adhesion protein | endo Co-IP | [55] | ||
| FAK | kinase | endo Co-IP | [56] | ||
| Filamin | actin binder | Co-IP | RZ | [57] | |
| GCKR | kinase | endo Co-IP | TD | [58] | |
| Gpx1 | peroxidase | Co-IP | TD | [59] | |
| GRA15 | virulence factor | Co-IP | [60] | ||
| GSTP1-1 | gluthation transferase | Co-IP | [61] | ||
| HGK | Kinase | Co-IP | [62] | ||
| Hoxa1 | transcription factor | THS, Co-IP | [63] | ||
| HSP70 | Chaperon | Co-IP | TD | [64] | |
| IKK1 | kinase | GST, endo Co-IP | RING | [65] | |
| IKK2 | kinase | GST, endo Co-IP | RING | [65] | |
| IKKe | kinase | Co-IP | [66] | ||
| IRE1 | kinase, nuclease | endo Co-IP | [67] | ||
| I-TRAF | adaptor | THS, GST, Co-IP | TD | [68] | |
| JIK | kinase | Co-IP | [45] | ||
| KRC | DNA binding | endo Co-IP | TD | [69] | |
| LGP2 | RLR | Co-IP | CTD | [70] | |
| LILRB3 | receptor | endo Co-IP | [71] | ||
| LRPPRC | RNA regulation | Co-IP | [72] | ||
| MAVS | adaptor | Co-IP | CTD | [73][74] | [73,74] |
| MEKK1 | kinase | Co-IP | [75] | ||
| MIZ | transcription factor | GST, endo Co-IP | RING | [76] | |
| MLK3 | kinase | Co-IP | NTD | [77][78] | [77,78] |
| MST1 | kinase | endo Co-IP | Zn fingers | [79] | |
| TRIM37 | E3 ligase | Co-IP | TD | [80] | |
| Nef | virulence factor | GST | [81][82] | [81,82] | |
| HCV core | virulence factor | GST | [81] | ||
| NIP45 | transcription factor associated | endo Co-IP | [83] | ||
| NSP1 | virulence factor | Co-IP | [84] | ||
| Nur77 | nuclear receptor | Co-IP | RING, NTD | [85] | |
| parkin | E3 ligase | endo Co-IP | [86] | ||
| proPTPRN2 | phosphatase | Co-IP | RING | [87] | |
| RET/PTC3 | oncogenic RTK fusion protein | Co-IP | [88] | ||
| RIPK1 | kinase | Co-IP | NTD, CTD | [89] | |
| RIP2 | kinase | Co-IP | [90] | ||
| RNAseT2 | ribonuclease | Co-IP | [91] | ||
| RSK2 | kinase | Co-IP | [92] | ||
| SHP-1 | phosphatase | Co-IP | [93] | ||
| SGEF | GEF | Co-IP | TD | [94] | |
| Sharpin | scaffold | Co-IP | [95] | ||
| SIAH-2 | E3 ligase | GST | [96] | ||
| SMAD4 | signaling protein | THS, endo Co-IP | [97] | ||
| SMURF-2 | E3 ligase | THS, Co-IP | [98] | ||
| SMYD2 | methyltransferase | Co-IP, SPR | [99] | ||
| SphK1 | kinase | GST, Co-IP | [100] | ||
| T2BP / TIFA | adaptor | THS, Co-IP | TD | [101] | |
| TCPTP | phosphatase | endo Co-IP | [102] | ||
| TPL2/COT1 | kinase | Co-IP | [103] | ||
| TAK1 | kinase | Co-IP | [104] | ||
| TBK1 | kinase | endo Co-IP | NTD | [74] | |
| TNIK | kinase | Co-IP | TD | [105] | |
| TRADD | adaptor | THS, Co-IP | CTD | [89][106] | [89,106] |
| TRAF1 | adaptor | THS, Co-IP | NTD, CTD | [2][89] | [2,89] |
| TRAF2 | E3 ligase, adaptor | THS, Co-IP | CTD | [2][89] | [2,89] |
| TRAF3 | E3 ligase, adaptor | Co-IP | [107] | ||
| TRAF4 | E3 ligase, adaptor | endo Co-IP | [108] | ||
| TRIF | adaptor | THS, Co-IP | [109] | ||
| UBC13 | E2 | endo Co-IP | RING | [76] | |
| USP4 | DUB | Co-IP | [110] | ||
| USP7 | DUB | GST | [80] | ||
| USP17 | DUB | Co-IP | [107] | ||
| UXT-V1 | transcriptional cofactor | endo Co-IP | [111] | ||
| VP4 | rotavirus capsid protein | THS, Co-IP | [112] | ||
| WDR62 | scaffold | Co-IP | [113] |
Abbreviations: Co-IP, co-immunoprecipitation of transiently expressed proteins; CTD, C-TRAF domain; endo Co-IP, co-immunoprecipitation of endogenous proteins; DUB, de-ubiquitinase; GST, glutathione-S transferase pull-down assay; NTD, N-TRAF domain; RING domain; TD, TRAF domain; THS, two-hybrid system; zinc, zinc finger domain.
The most important TRAF2-interacting E3 ligases are cIAP1 and cIAP2. Prominent substrates of the TRAF2-cIAP1/2 complex are kinases, such as transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), NFκB-inducing kinase (NIK), apoptosis signal-regulating kinase 1 (ASK1), and receptor-interacting kinase 1(RIPK1), which regulate NFκB signaling and induction of programmed cell death. Intriguingly, in some special cases TRAF2 may also act itself as an E3 ligase by help of its RING domain, such as in context of TRAIL death receptor signaling where TRAF2 K48-ubiquitinates caspase-8 [114][115][114,115]. Like most other TRAF family members, TRAF2 is involved in the engagement of signaling pathways resulting in the activation of transcription factors, such as the two NFκB pathways, various mitogen-activated protein (MAP) kinase cascades, and the MAVS/TBK1/IRF3 pathway. However, TRAF2 can also affect cellular functions independent from transcription-stimulating pathways by triggering phosphorylation and/or ubiquitination of proteins, thereby regulating their activity, stability, function, or localization. Examples therefore are K48-ubiquitiantion and proteasomal degradation of caspase-8, cRel, interferon regulatory factor 5 (IRF5), and unc-51-like autophagy activating kinase 1 (ULK1) triggered alone by TRAF2 (caspase-8) or by TRAF2 in concert with TRAF3 and cIAP1 and cIAP2 (cRel, IRF5, ULK1) [114][115][116][117][114,115,116,117]. Further examples of “transcription”-independent TRAF2 activities are the engagement of the Src homology 3 domain-containing guanine nucleotide exchange factor (SGEF), leading to glioblastoma cell migration in response to Fn14 activation [94], and K63-ubiquitination of dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A), promoting its translocation to vesicles to attenuate epidermal growth factor receptor (EGFR) degradation [51] and its role in mitophagy [85][118][119][85,118,119].
2. Role of TRAF2 in Immune Signaling Pathways
2.1. TRAF2 and Activation of the Classical NFκB Pathway
Nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB) are homo- and heterodimeric transcription factors formed of the five NFκB proteins p65/RelA, RelB, cRel, p50, and p52, of which the latter two are initially expressed in the form of precursor molecules (p100 and p105). NFκB dimers are held in check by cytoplasmic retention [120][121][120,121] resulting from the fact that the nuclear localization sequence (NLS) of NFκBs is blocked in non-stimulated cells by either of two related mechanisms. First, by forming a ternary complex with ankyrin-repeat containing inhibitor of κB proteins (IκBs), e.g., IκBα, or second, by incomplete maturation of the precursor proteins p100 and p105 containing a C-terminal autoinhibitory ankyrin-repeat domain. There are two distinct signaling mechanisms that relieve the NLS of NFκBs from the inhibitory interaction with ankyrin repeats: firstly, the IκB kinase (IKK) complex-induced degradation of IκB proteins and the IKK-induced processing of p105 (classcial or canonical NFκB pathway) and secondly, the NIK-induced processing of p100 to p52 (alternative or non-canonical NFκB pathway) (Figure 2).
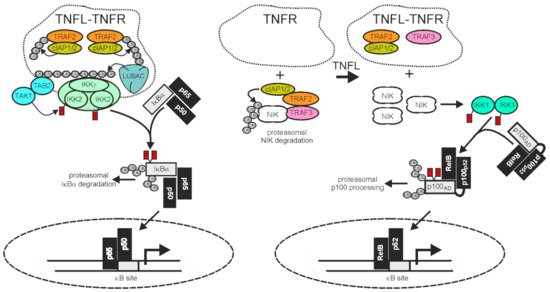
Figure 2. TRAF2 and the cIAPs in receptor-induced activation of the classical and alternative NFκB pathway. The activities of TRAF2 and the cIAPs have opposing qualities in the classical (left panel) and alternative (right panel) NFκB signaling pathway. In the classical NFκB pathway TRAF2 and the cIAPs enable signaling, while in the alternative NFκB pathway they act as inhibitors. Importantly, TNFR-induced recruitment of TRAF2 and the cIAPs, which triggers the classical NFκB pathway, is linked with an “inhibitory” relocation of these molecules away from their cytosolic substrate NIK in the alternative NFκB pathway. Therefore, TNFRs eventually stimulate both NFκB signaling pathways despite the opposing quality they have in these pathways. For more details, refer to main text.
A major function of TRAF2 is to transduce activating signals from cell surface receptors, particularly TNFRs, to the IKK complex in the classical NFκB pathway. The latter phosphorylates IκBα and related IκBs to trigger their proteasomal degradation, the key event in activation of the classical NFκB pathway (Figure 2). To fulfill its tasks in classical NFκB signaling, TRAF2 directly or indirectly recruits to the liganded receptor molecules along with the TRAF2-interacting E3 ligases cIAP1 and cIAP2. This results in the activation of the latter. The cIAPs in turn K63 ubiquitinate TRAF2 and other proteins present in the receptor signaling complexes, and thereby create docking sites facilitating the recruitment of the linear ubiquitin assembly complex (LUBAC). The latter catalyzes the formation of linear M1-linked ubiquitin chains, creating binding sites for the NFκB essential modulator (NEMO), a subunit of the IKK complex, and the TAK1-binding-protein-2 (TAB2) subunit of the IKK-engaging TAB2-TAK1 complex. Worth mentioning, TRAF2 also triggers the recruitment of regulatory proteins, such as deubiquitinases, that terminate/resolve the ubiquitination events leading to IKK activation. For example, the cylindromatosis tumor suppressor (Cyld) directly interacts with TRAF2 and removes K63-linked polyubiquitin chains from TRAF2, resulting in reduced NFκB signaling and enhanced apoptosis but also in the maintenance of hematopoietic stem cell dormancy by inhibition of p38 MAP kinase signaling [49][122][123][124][49,122,123,124]. Likewise, A20 [also named TNFα-induced protein 3 (TNFAIP3)] acts as a K63 deubiquitinase, e.g., for RIPK1, NEMO/IKKγ, or caspase-8, but also, alone or in concert with other E3 ligases, as a K48 E3 ligase [125]. In accordance with the function of its major substrates and in view of the fact that A20 itself is a NFκB target, A20 has been implicated in the downregulation of the classical NFκB pathway and the control of cell death [125].
The generalized mechanisms of receptor-induced TRAF2-mediated activation of the IKK complex described above have been primarily investigated for TNFR1, but there is evidence that similar or related mechanisms also apply for other receptors. For example, cIAPs and/or the LUBAC have also been implicated in NFκB activation by other TRAF2-utilizing receptors, such as CD40, TNFR2, Fn14, and the TRAIL death receptors [126][127][128][129][126,127,128,129]. Intriguingly, the NFκB-inhibitory effects of dominant-negative TRAF2 mutants on TNFR signaling reported in early years is often more pronounced than the inhibitory effect observed in receptor stimulated TRAF2-deficient cells. A possible explanation for this is that other TRAF proteins, which use overlapping binding sites to TRAF2 in the considered TNFR type, act redundantly with TRAF2 and/or fulfill functions distinct of those of TRAF2. In fact, there is evidence that TRAF2 and TRAF5 act redundantly in TNF-induced classical NFκB signaling and that TRAF2, in cooperation with TRAF1 and TRAF6, redundantly signal CD40-induced NFκB activation [130][131][130,131]. Furthermore, it is well-established that TRAF2 cooperates with TRAF3 in the control of alternative NFκB signaling (see 2.2.). In general, however, redundancy and/or cooperativity between TRAF2 and other TRAF proteins have been limitedly investigated so far.
2.2. TRAF2 and Activation of the Alternative NFκB Pathway
TRAF2 and the cIAPs play a central role in the control of the alternative NFκB signaling pathway. In the cytoplasm TRAF2 interacts via TRAF3 with NIK which is constitutively active and enables cIAP1/2-mediated K48-ubiquitination of the latter, resulting in its proteasomal degradation [132][133][132,133]. NIK activates IKKα which in turn phosphorylates p100, triggering its proteasomal processing to p52. Therefore, TRAF2, TRAF3, and the cIAPs finally inhibit the alternative NFκB pathway. Thus, in the classical NFκB pathway, TRAF2 and the cIAPs trigger the degradation of pathway inhibitory ankyrin-repeat proteins or ankyrin-repeat domains (IκBs, ankyrin domain of p105), whereas in the alternative NFκB pathway, the same proteins prevent, together with TRAF3, the degradation of a pathway inhibitory ankyrin-repeat domain (Figure 2). The alternative NFκB pathway is typically engaged by members of the TNFRSF, such as Fn14, CD40, TNFR2, and the LTβR. In view of the opposing effects of TRAF2 and the cIAPs on ankyrin-repeat containing NFκB-inhibitory proteins/protein domains in the two NFκB signaling pathways, it first seems counter-intuitive that activation of TNFRs results in the concomitant activation of both pathways. However, this apparent contradiction is resolved when two points are considered: i) that the amount of cell-expressed TRAF2 and cIAP1/2 molecules is limited and ii) that TRAF2 and the cIAPs, along with TRAF3, act constitutively in the cytoplasm of unstimulated cells in context of the alternative NFκB pathway but fulfill their role in the classical NFκB pathway in an inducible manner in plasma membrane-associated receptor signaling complexes. Ligand-induced recruitment of TRAF2 (and/or TRAF3) and the cIAPs to plasma membrane-receptors is accordingly intimately linked to the depletion of these molecules from the cytosol, resulting not only in the formation of classical NFκB-stimulating receptor complexes but also in a reduction of the cytosolic available amount of TRAF2-cIAP1/2 complexes that can be recruited via TRAF3 to NIK to inhibit the alternative NFκB pathway. It is worth mentioning that the sole depletion of TRAF2, TRAF3, and the cIAPs from the cytoplasm is sufficient to engage the alternative NFκB pathway [132][133][134][135][132,133,134,135] but that this mechanism can be enhanced in its effects by receptor-associated degradation of the TRAFs and the cIAPs. Taken together, despite the opposing quality of the activity of TRAF2 and the cIAPs on the two NFκB signaling pathways, ligand-induced receptor-TRAF2 interaction eventually results in concomitant activation of both pathways.
2.3. TRAF2 in RLR Signaling
In RNA virus-infected cells cytosolic double-stranded (ds) RNA is recognized by RIG1 and/or the RIG1-like receptor (RLR) melanoma differentiation-associated 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2) [136]. Binding of dsRNA by RIG1 and MDA5 enables these proteins to convert from an autoinhibited form to a tetrameric “open form” which, assisted by the E3 ligase RIPLET and K63-polyubiquitination, assembles into filaments [137][138][139][137,138,139] (Figure 3).
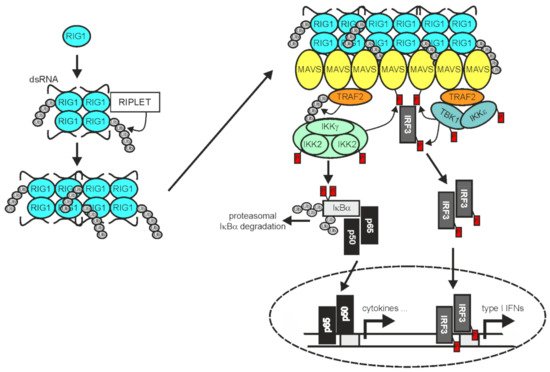
Figure 3. TRAF2 in RIG1 signaling. Binding of dsRNA by RIG1, results in conformational change, K63-ubiquitination, and filament formation. RIG1 filaments in turn instruct filament formation of mitochondria-associated MAVS. The MAVS filaments enable recruitment of TRAF2 and IRF3, but also other TRAF proteins not indicated here. TRAF2 and the other TRAF proteins mediate the recruitment of the IKK complex and TANK-binding kinase 1 (TBK1)/IKKε enabling activation of the classical NFκB pathway and IRF3 by the mechanisms described in detail in the text. Please note, TRAF2 acts independently here from cIAP1 and cIAP2 [74]. For more details refer to main text.
The RLR filaments in turn bind to mitochondria antiviral signaling protein (MAVS; also named VISA, IPS-1, or Cardif) and nucleate the formation MAVS filaments [140]. The latter in turn act as signaling platforms, like aggregated TNFRs, to recruit TRAF2, TRAF3, TRAF5, and TRAF6 along with IRF3 and the TRAF-interacting IKK- and TBK1/IKKε complexes to engage downstream signaling pathways, namely the classical NFκB pathway and the TBK1/IRF3/IFNβ pathway [74][141][142][74,141,142]. TRAF2 and TRAF5 on the one side and TRAF3 and TRAF6 on the other side bind to different binding motifs in MAVS and act redundantly to activate a strong innate immune response [141]. While the RING domains of TRAF2 and the other TRAFs were found to be important to mediate NEMO ubiquitination and IKK activation in context of RLR signaling, they appeared dispensable for activation of the TBK1-IRF3 axis [74]. In contrast to receptors of the TNFRSF, RIG1 and MDA5 not only stimulate the activation of NFκB transcription factors by help of the TRAF proteins but also engage IRF3 and IRF7. The reasons for the different signaling qualities of TRAF2 and the other TRAFs in TNFR versus RLR signaling are still unclear. It is worth mentioning that the RLR LGP2 associates with the C-terminus of TRAF2, TRAF3, TRAF5, and TRAF6 and acts as a pan-inhibitor of stimuli using these TRAF proteins for activation of the classical NFκB pathway [70].