Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Daniel R. Halloran and Version 2 by Catherine Yang.
Protein kinase CK2 (CK2) is a ubiquitous holoenzyme involved in a wide array of developmental processes. The involvement of CK2 in events such as neurogenesis, cardiogenesis, skeletogenesis, and spermatogenesis is essential for the viability of almost all organisms, and its role has been conserved throughout evolution.
- casein kinase II
- development
- cancer
1. CK2 and Development
CK2 is highly expressed and prominent throughout many stages of development. During early embryogenesis, CK2 activity was identified first at embryonic day 11 (E11) in mice, rats, and chickens [1][2][3][26,27,28]. Continuously after this day, CK2 regulates the formation of various organ systems. Specifically, during neurogenesis and regarding the negative regulation of neuron differentiation, CK2 phosphorylates Groucho/Transducin-like enhancer (TLE1) to limit the formation of neurons [4][29]. CK2α null mice display improper neural tube formation, emphasizing the essential role of CK2 in this process [5][30]. Furthermore, CK2 contributes to memory formation, and the overexpression of this kinase is correlated with improved long-term memory [6][31]. In cardiogenesis, CK2, specifically CK2α’, is essential for the proper formation of the heart, and knockouts of this subunit lead to irregular cardiogenesis, as well as death in mid-gestation [5][30]. Similarly, CK2 is critical for proper spermatogenesis and the fertility of organisms [7][32]. While CK2α’−/− mice remain viable, male mice are infertile and incapable of producing offspring, as germ cells do not form properly [5][8][9][30,33,34]. Moreover, CK2α’ is highly expressed in testis, and a lack of CK2α’ also leads to oligozoospermia [8][33]. CK2 is also required for the formation of the skeleton during development, specifically at E17.5 [10][35]. It has also been demonstrated that the conditional deletion of the gene encoding beta subunit of CK2 (Csnk2b) leads to the shortening of the limbs, improper endochondral ossification, and lethality in mice [10][35]. Therefore, CK2 is critical for many stages of development and the production of viable organisms (Figure 1).
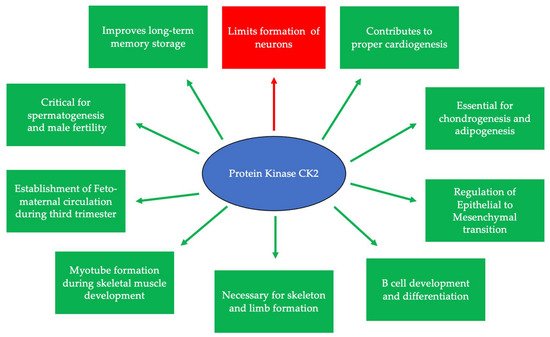
Figure 1. CK2 is expressed at embryonic day 11 and is critical for many developmental processes. Specifically, CK2 is necessary for limiting neurogenesis and preventing the excessive differentiation of neurons. In addition, CK2 expression promotes long-term memory storage. Further, this protein is essential for skeletogenesis, chondrogenesis, adipogenesis, and proper limb formation. CK2 contributes to spermatogenesis, and the inhibition of its expression leads to infertility. Finally, CK2 is important for B cell differentiation and development, myotube formation, the regulation of the epithelial-to-mesenchymal transition (EMT), and the establishment of circulation between the fetus and the mother during the third trimester.
There are three classifications of substrates for CK2. Class I substrates are equally phosphorylated by the holoenzyme and free catalytic subunits, whereas Class II substrates are phosphorylated by only free catalytic subunits but not by the holoenzyme. Further, substrates of Class III are phosphorylated preferentially by the holoenzyme but not by free catalytic subunits. Thus, the phosphorylation of these different classes varies during development (Figure 2) [11][45]. To study the role of the individual subunits during development, specific subunit knockouts were induced.
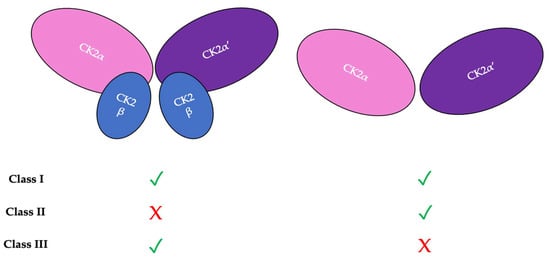
Figure 2. There are three classifications of CK2 substrates. Class I substrates are identified as proteins that are equally phosphorylated by the holoenzyme and individually by the catalytic subunits. Class II substrates are specifically phosphorylated by the catalytic submits of CK2 but not by the holoenzyme. Class III substrates are preferentially targeted and phosphorylated by the holoenzyme but not by the catalytic subunits of CK2.
2. CK2 Expression and the Progression of Diseases
The ubiquitous expression of CK2 occurs within almost all cellular compartments [12][128]. The continued identification of its newer substrates broadens the understanding of the processes in which this protein is involved [13][129]. Alternative forms of this holoenzyme, such as single catalytic or regulatory subunits, have differential expression and functions within tissues. The CK2α’ subunit has enhanced mRNA expression in the testis, skin, and across brain tissue. Further, the CKα and CK2β subunits have a more ubiquitous expression within tissues [14][130]. Oligomeric super-structures of the CK2 holoenzyme also exist, in which this protein controls the exposure of the catalytic active sites and subsequent enzymatic activity [15][131]. The balance between the number of catalytic CK2α and regulatory CK2β subunits is maintained by a mechanism whereby CK2α activates the transcription factors for CK2β when its levels are depleted. Once sufficient levels of CK2β are obtained, transcription is inhibited by holoenzyme formation, which then disables the interaction between CK2α and its own transcription factors [16][132].
2.1. Dynamic Localization of CK2
The subcellular localization of CK2 determines its regulation, which predominantly includes the cytoplasm, plasma membrane, and nucleus. Interactor proteins and substrates of CK2 are located predominantly in these three cellular compartments [17][140]. The localization of CK2 varies during cell cycle progression and is sensitive to external stimuli, such as radiation, hypoxia, and stress. Exposure to ionizing radiation (IR) was identified to alter CK2α subunit localization from the cytoplasm to the nucleus in human non-small cell lung cancer cell lines, including H460, A549, and PC9 (Figure 3). The nuclear localization is reported to be transient. In addition, there is an increase in the kinase activity of CK2 during the S phase when cells are exposed to IR. Furthermore, with the pharmacological inhibition of CK2, there is an increase in IR sensitivity [18][19][141,142]. This emphasizes the role of CK2 in transmitting radiation stimuli to the nucleus and its involvement in the DNA damage response. Similarly, in M059K human glioma cells, it colocalizes with the IR-induced DNA damage sites [20][108]. Resistance to IR treatment could be attributed to the dynamic localization of this kinase. During hypoxia, CK2 activity is seen to increase without an increase in its expression, but rather by the transport of catalytic α and α’ subunits to the nucleus (Figure 4).
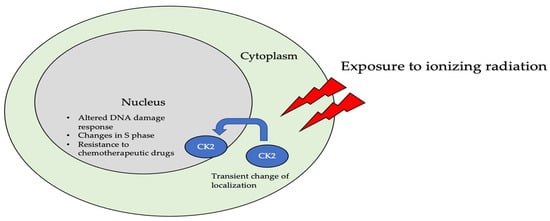
Figure 3. The localization of CK2 changes when cells are exposed to ionizing radiation (IR). Specifically, CK2α is translocated from the cytoplasm to the nucleus in multiple cell lines, including A549, H460, PC9, and M059K. Further, upon radiation exposure, the overall kinase activity of CK2 increases, emphasizing its role in the DNA-damage-repair response, as it colocalizes with DNA in the nucleus of cells.
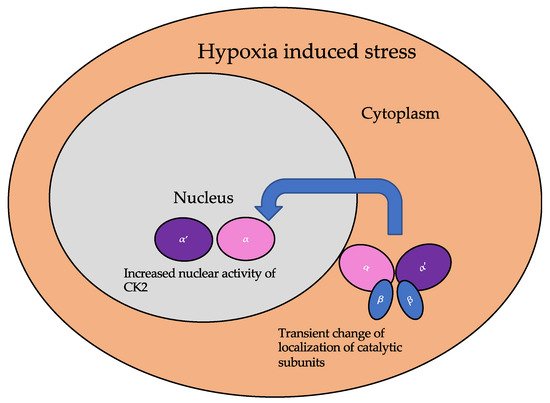
Figure 4. The localization of the catalytic subunits of CK2 is altered during hypoxia. Specifically, these subunits are translocated to the nucleus transiently to aid the cellular response to hypoxia.
2.2. CK2 in Diseases
CK2 is associated with the etiology of many diseases due to its pleiotropic nature and involvement in almost all cellular process. The diseases associated with the aberrant expression or function of this kinase include cancer, multiple sclerosis, cystic fibrosis, diabetes mellitus, neurological disorders, cardiac hypertrophy, and inflammation [21][102]. The examples delineated below describe the involvement of CK2 in disease progression based on unique mechanisms specific for the disease. The implication of aberrant CK2 expression and/or function is apparent in several studies.
2.2.1. CK2 in Cancer Progression
CK2 regulates the progression of at least 15 cancer-related proteins including the tumor suppressor p53, histone-modification enzymes such as HDAC1 and HDAC2, and NF-κB subunit RelA [22][146]. The interaction of additional direct interactors and indirectly regulated proteins remains unclear. There is an upregulation of CK2 during the tumor progression of many cancers and leukemia [23][24][147,148]. CK2 itself can be used as a prognostic marker for some cancer types[25][149]. Proteomic analyses of cell lines indicates that the catalytic CK2α subunit is predominantly expressed in several cancerous cell lines, including U2OS from human osteosarcoma, epidermoid squamous cell carcinoma, brain glioblastoma A549 from lung carcinoma, GAMG from glioblastoma, HEK293 from embryonic kidney cells, HeLa from cervical carcinoma, HepG2 from hepatoma, Jurkat from
As CK2 regulates the activation of several proteins required for the cell cycle, the consequences of its overexpression are exacerbated in solid tumors and leukemias [26][165]. CK2 overexpression and/or overactivity results in the abnormal phosphorylation of its targets. One example is tumor suppressor Ikaros, which is a transcriptional repressor of the genes encoding for Bcl-2-like protein 1 (BCL2L1, PIK3CD, and PIKFYVE) [27][28][98,100].
The central role of CK2 in pathway networks for cell cycle regulation and proliferation makes this protein an interesting pharmacological target for treatment of cancer.
2.2.2. CK2 and Diabetes and Obesity
In pancreatic β cells, a G-protein coupled-receptor muscarinic 3 receptor (M3R) regulates insulin release and homeostasis. CK2 is a negative modulator of M3R. Its phosphorylation of the M3Rs on β-cells inhibits insulin release [29][171]. Therefore, by phosphorylating the M3Rs, CK2 prevents the release of insulin. However, the abnormal activity of CK2 is linked to diabetes due to its role with these receptors.
Sirtuin1 (SIRT1) deacetylase is a substrate of CK2 and is a regulator for hepatic lipid metabolism. This protein modulates transcription as an energy sensor molecule with its deacetylase function. In diet-induced obesity, CK2 is highly overexpressed and therefore increases the activity of SIRT1. Aberrant phosphorylation of SIRT1 at serine 164 by CK2 inhibits the entry of the former into the nucleus and mildly impairs deacetylase function, thus hampering its regulatory function in hepatocytes, and has been linked to the onset of obesity [30][172].
2.2.3. CK2 and Heterotrophic Bone Formation
Heterotrophic bone formation in soft tissue due to injury, medication, or as a symptom of another disease can be a painful condition. CK2 phosphorylation stabilizes runt-related transcription factor 2 (RUNX2) by deubiquitylation through the CK2/herpesvirus-associated ubiquitin-specific protease (HAUSP)/RUNX2 pathway. This regulation is important during skeletal development for the fine-tuning of RUNX2, which is a master transcription factor during osteoblastogenesis. However, the overexpression or overactivity of CK2 during heterotrophic bone formation causes ectopic osteoblastogenesis from skeletal stem cells by the same mechanism for RUNX2 stabilization [10][35].
2.2.4. CK2 and Cardiovascular Diseases
Within the cardiovascular system, CK2α interacts with p27 to prevent its ubiquitination or degradation. Here, the stabilization of p27, which is an inhibitor of cell progression, leads to its accumulation in cytoplasm and the subsequent apoptosis of the cell. Further, CK2α is responsive to external growth stimuli to regulate its interaction with p27. The interaction between CK2α and p27 is interrupted during cardiac hypertrophy [31][173]. Here, transcriptional reprogramming leading to fetal gene expression causes cardiac hypertrophy. In addition, this reprogramming is related to HDAC2 activity. Specifically, CK2α phosphorylates HDAC2 at serine 394, causing its activation, and leads to cardiac hypertrophy [32][174].
Patients with cardiac desynchrony are associated with higher risk for morbidity and mortality from heart failure compared to patients with synchronic heart function. Cardiac resynchronization therapy (CRT, pacemaker) is the only known non-pharmaceutical intervention that has long-term effects on this disorder. However, some patients do not respond to this intervention. To study molecular mechanisms for dyssynchronous heart failure (HFdys) and the impact of CRT, proteomic analysis was performed. The comparison of phosphoproteome of HFdys before and after CRT led to the discovery that CK2 signaling is activated during HFdys and that CRT reverses it [33][175]. The contribution of CK2 signaling in cardiac desynchrony is still an ongoing area of research.
2.2.5. CK2 and Neurodegenerative Disorders
The expression and activity of CK2 in brain cells is involved in neurodegenerative disorders [34][176]. Here, the overexpression of CK2 is associated with neurodegeneration [35][177]. Further, there are numerous CK2 substrates associated with brain development and function. In Alzheimer’s disease, CK2 colocalizes with the neurofibrillary tangles. The hyperphosphorylation of the SET protein at serine 9 leads to its improper localization in the cytoplasm caused by the activation of CK2 by tau or β-amyloid (Aβ). The overexpression of CK2 also causes cognitive deficits by impairing synaptic plasticity and synaptogenesis [36][178]. In Parkinson’s disease (PD), CK2 is localized to Lewy bodies and phosphorylates α-synuclein and synphilin-1 [37][179]. The treatment of PD using levodopa (L-DOPA) is known to induce L-DOPA-induced dyskinesis (LID). CK2 is important for the regulation of pathways leading to LID. Specifically, CK2α downregulation reduces the severity of LID [34][176].
Regarding dopaminergic signaling, CK2 is a negative regulator of the D1 receptor (D1R). It binds directly to the Gαs or Gαolf (Golf) subunits, and its knockout leads to the elevated expression of D1R on the plasma membrane. This leads to an increased response to D1 agonists [38][40]. With the CK2 conditional knockouts in medium spiny neurons, which express dopaminergic receptors, CK2α regulates the D1 signaling pathway in vivo [39][180].
2.2.6. CK2 and Neurological Disorders
Further, in the neuropsychiatric disorder Tourette syndrome (TS), a mutation in the SLITRK1 gene is present. The gene product is a membrane-bound protein that regulates synapse formation. The symptoms of TS arise early in the childhood, and affected individuals often suffer from attention-deficit/hyperactivity disorder (ADHD) and obsessive–compulsive disorder (OCD). CK2 phosphorylates the SLITRK1 protein at its 14-3-3 binding site, and a mutation at this phosphorylation site inhibits neurite formation [40][181].
2.2.7. CK2 in Infectious Diseases
Viral proteins are substrates of CK2. The presence of host kinase phosphorylation sites within these proteins can be useful for the virus to identify host cell status [41][182]. In the proteome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a total of 25 phosphorylation sites have been identified. CK2 is one of the main kinases identified to regulate these sites. There is also activation of CK2 signaling observed during SARS-CoV-2 infection [41][182].
The hepatitis C virus (HCV) nonstructural protein 5A (NS5A) plays an important role in viral particle assembly. Structural studies of the NS5A and CK2 complex using time-resolved nuclear magnetic resonance (NMR) indicate that there are four possible CK2 phosphorylation sites in the NS5A D3 domain [42][183]. The expression of hepatitis B virus (HBV) is regulated by the Human La protein (hLa). The phosphorylation of hLa at serine 366 by CK2 activates the protein and increases HBV expression. The pharmacological inhibition of CK2 by tetrabromobenzimidazole (TBBz) or a knockdown its gene results in a reduction of HBV expression [43][184].
In prion diseases, the pathogenic form of the prion protein (Prp) interferes with cellular processes, such as fast axonal transport (FAT), which causes an onset of neurodegenerative disorders. The intracellular accumulation of cellular prion protein (PrPC) and full-length PrP (PrP-FL) induces neuronal toxicity, causing severe ataxia in mice, and further, FAT is often inhibited. CK2 inhibits FAT in a similar pattern as in PrP-FL-induced symptoms [44][45][46][47][48,185,186,187].
2.2.8. Regulation of Immune Response by CK2
The regulation of the immune cell response is targeted for the use of CK2 inhibitors. However, delineating its role during development in the immune response under disease progression has been a challenge. The involvement of CK2 is being investigated during this disease response in ongoing studies.
During Listeria monocytogenes (Lm) infection, the adaptive and innate immune responses are involved. In myeloid cell-specific conditional knockout of CK2α in mice, host resistance to Lm infection is significantly increased without affecting myeloid cell development. Myeloid cell recruitment is also seen to be negatively regulated by CK2α [48][188].
Further, the expression of CK2 is increased in the inflamed mucosa of ulcerative colitis (UC) patients. It is important for maintaining reciprocal balance between Th17 and Treg cells [49][189]. Interestingly, CK2 activity is reduced, along with an increase in reactive oxygen species (ROS) generation, during acute colitis. NADPH oxidase 1 (NOX1), which is an important regulator of mucosal immunity, is dysregulated during inflammatory bowel disease. Further, CK2 is a suppressor of NOX1 activity [50][123].
2.2.9. CK2 and Senescence
CK2 activity and expression is downregulated in older animals. Further, the pharmacological inhibition of CK2 induces cellular senescence [51][52][190,191]. With artificial downregulation of CK2 by the activity of histone trimethylases, such as histone-lysine N-methyltransferase SUV39H1(SUV39h1), its activity is upregulated in senescence and leads to an increase in tri-methylation at the 9th lysine residue of the histone H3 (H3K9me3) and senescence-associated heterochromatin formation (SAHF). An increase in SAHF leads to the suppression of genes associated with cell cycle progression, such as cyclin D1 [53][192].
Next, in CK2α knockdown cell lines (MCF-7 and HCT116 cells), the expression of histone demethylases [JmjC domain-containing histone demethylase (JMJD2/KDM4) and lysine-specific demethylase 1 (LSD1/KDM1a)] is downregulated at the translational level. The ectopic expression of these histone demethylases suppressed SAHF and senescence-associated β-galactosidase activity. The effects of the translational downregulation of LSD1 due to the reduction in CK2 activity are complex, since it has non-histone substrates such as p53.
3. Clinical Applications of CK2
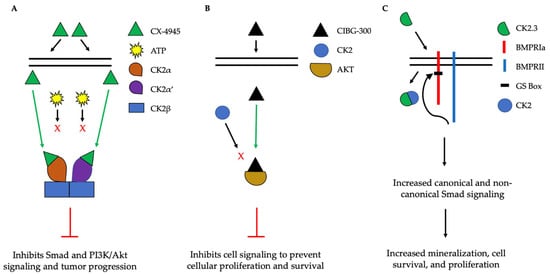
The implication of CK2 in the progression of many diseases has led to its popularity as a therapeutic target. Specifically, researchers have attempted to prevent the kinase activity of CK2, thereby to prevent pathological activity. In cells, CK2 contains pocket domains for adenosine triphosphate (ATP)-binding [54][195]. The pocket domain is essential for the activation and subsequent enzymatic activity of CK2. Therefore, blocking this domain may deactivate CK2. One such inhibitor under investigation is CX-4945, known as silmitasertib in the clinic. CX-4945 is currently in clinical trials to treat cancer and is being tested as an oral administration to patients to limit the activity of CK2. Specifically, CX-4945 functions as an ATP-competitive inhibitor, leading to cell cycle arrest and apoptosis [55][196]. Here, CX-4945 inhibits the activity of both CK2α and CK2α’ by binding to the ATP-binding site of these subunits with a higher affinity than ATP [54][56][57][58][195,197,198,199]. This competitive binding leads to the decreased expression of CK2 and its subsequent signaling, such as Smad and PI3K/Akt pathways [55][58][196,199] (Figure 5A). This decreased activation of these pathways by CX-4945 has inhibited the progressions of several cancers, including gastric cancer, renal cancer, hematological cancer, cholangiocarcinoma, basal cell carcinoma, and medulla blastoma [56][59][60][61][24,197,200,201]. CX-4945 is a promising therapeutic agent and has displayed minimal side-effects, such as diarrhea, nausea, and anemia, suggesting its role as a safe treatment [61][62][201,202].
Figure 5. Simplified schematic demonstrating three therapeutics targeting the activity of CK2. (A). CX-4945 is a competitive ATP inhibitor that binds preferentially to ATP-pocket domains. Here, it binds to both CK2α and CK2α’, thereby preventing ATP from binding and activating CK2. CX-4945 has prevented tumor progression by inhibiting signaling pathways such as Smad and PI3K/AKT. (B). CIBG-300 functions by binding to conserved phosphorylation sequences on the substrates of CK2, such as Akt. This association prevents the phosphorylation of these substrates by CK2, leading to decreased cell signaling, proliferation, and survival. (C). CK2.3 is uptaken by cells and binds to CK2, therefore preventing its association with BMPRIa. As a result, BMPRII can phosphorylate BMPRIa, leading to the downstream activation of signaling pathways that induce mineralization and cell survival.
Another CK2 inhibitor, known as CIBG-300, is also currently in clinical trials as a therapeutic to treat cancer [63][64][106,203]. This inhibitor binds specifically to CK2 to prevent cellular proliferation; leads to decreased cell adhesion and migration; and promotes the apoptosis of the cells, specifically in cervical cancer, breast cancer, and colorectal cancer, both in vivo and in vitro [64][65][66][67][68][69][70][71][203,204,205,206,207,208,209,210]. Here, CIBG-300 functions by binding to the conserved phosphorylation sequences of CK2 substrates [68][72][207,211]. Subsequently, CK2 is unable to phosphorylate its downstream target; CIBG-300 thereby prevents the activity of various proteins involved in cancer progression and inhibits angiogenesis [68][69][70][73][74][207,208,209,212,213]. More interestingly, CIBG-300 inhibits the activation of Akt, PI3K, PTEN, and NF-κB signaling pathways, therefore decreasing cell proliferation and survival [75][76][77][95,214,215] (Figure 5B). Further, CIBG-300 treatment leads to limited side-effects including edema, hot flashes, tachycardia, lower abdominal pain, and bleeding [74][213]. Thus, this potential therapeutic has been efficient in clinical trials and holds promise to treat a wide array of cancers.