Oral papilloma lesions may appear as a result of HPV infection, or not, and only special molecular methods could differentiate them. Low-risk and high-risk HPV types could induce oral HPV papillomatosis with different natural evolution, clearance and persistence mechanisms. The pathogenic mechanisms are based on the crosstalk between the oral epithelial and immune cells and this very efficient virus. HPV acts as a direct inducer in the process of transforming a benign lesion into a malignant one, the cancerization process being also debated in this paper. According to the degree of malignity, three types of papillomatous lesions can be described in the oral cavity: benign lesions, potential malign disorders and malignant lesions. The precise molecular diagnostic is important to identify the presence of various virus types and also the virus products responsible for its oncogenicity. An accurate diagnostic of oral papilloma can be established through a good knowledge of etiological and epidemiological factors, clinical examination and laboratory tests.
- oral papilloma
- oral HPV infection
- HPV-related oral lesions
- HPV immunity
- HPV oncogenicity
1. Introduction
2. HPV Morphology
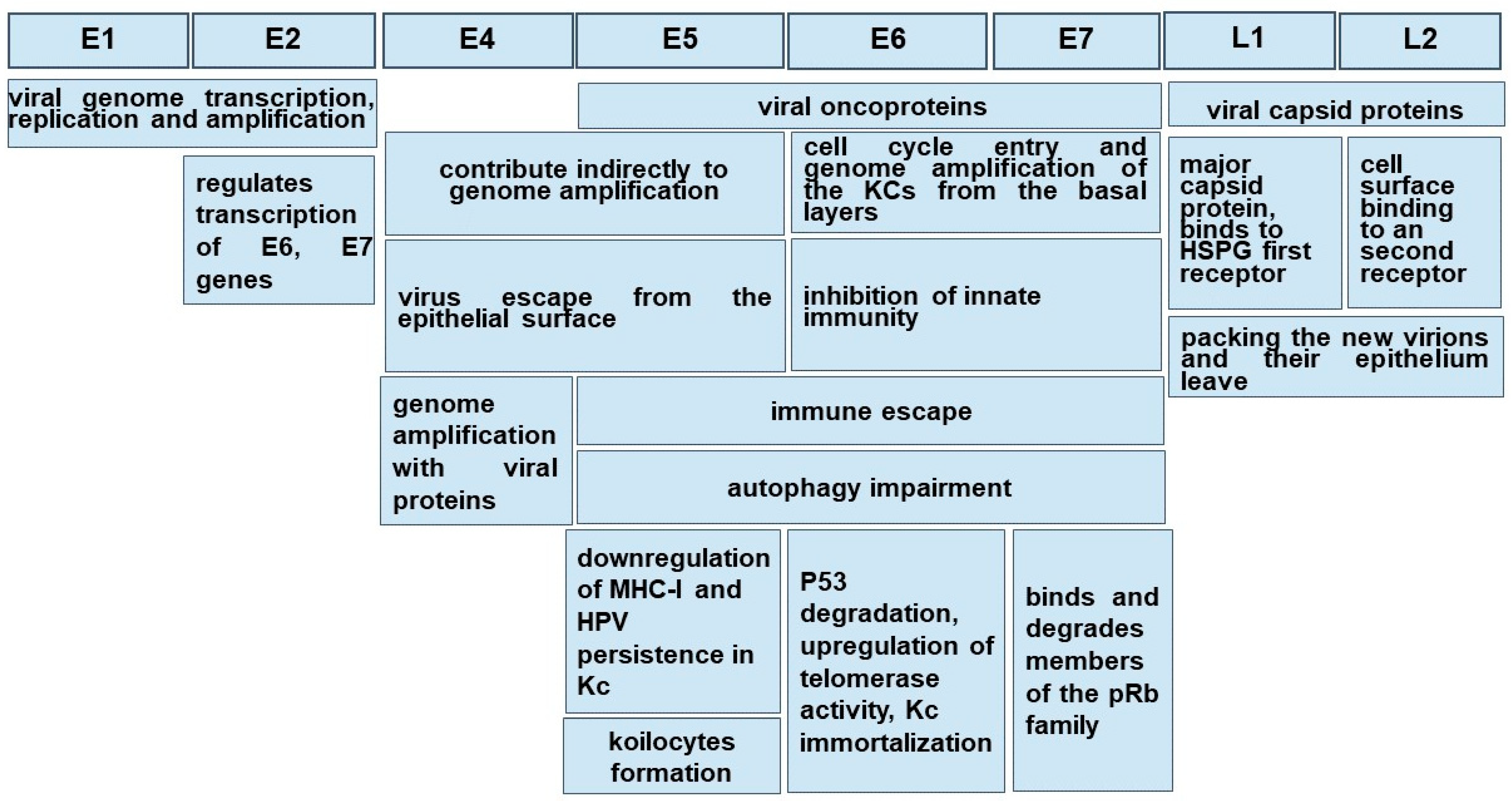
Morphologically, these non-enveloped viruses have a hemispherical shape and dimensions between 50 and 60 nm [2,29,30][2][29][30]. Even in a small virus of 8 Kpb, the HPVs are composed of eight genes, called open reading frames (ORF), six early (E1, E2, E4, E5, E6 and E7) and two late (L1 and L2), necessary for different stages of the virus cycle, and a non-coding long control region (LCR). The LCR contains a DNA replication origin and binding sites for host cell transcription factors and for the viral E1 and E2 proteins that control viral replication and gene expression. E5, E6, and E7 encode three viral oncoproteins, E6 and E7 being the dominant oncoproteins of high-risk HPVs that modulate the transformation process [1,2,29][1][2][29]. The morphology of a high-risk HPV is displayed in Figure 1.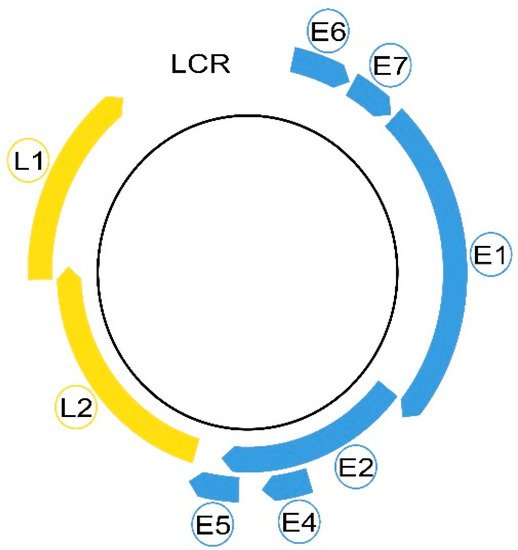
3. HPV Transmission
HPV infection of the oral mucosa is determined by sexual or non-sexual transmission. The oro-sexual practices are incriminated in the sexual transmission, correlated with sexual behavior (number of partners, age of sexual live onset [39,40][39][40]). Interestingly, a horizontal transmission via saliva from mouth to mouth is also possible, as oral sex is not compulsory for HPV infection from a positive partner [1]. It seems that the bifocal transmission of oral and vaginal mucosa between partners is not usual, because they have different susceptibility, and the protective attribute of the saliva may be considered [39]. Vertical transmission is also accepted, the newborns with HPV-positive mothers having 33% more risk for HPV infection [1]. Oral papillomatous lesions can be transmitted from the skin to the oral mucosa. An eloquent example of this situation is verruca vulgaris, frequently found in children [40,41][40][41].4. HPV Natural Infection
Both low-risk and high-risk HPVs have an affinity for the squamous epithelium of the oral and cervical mucosa and infect epithelial cells (Kcs). HPV’s capacity to immortalize in vitro in oral Kcs allows the extrapolation of the biological infection from the genital mucosa to the oral one [39]. The mucosa lining the oral cavity is formed of an epithelium sustained by a basal membrane (BM) which joins the epithelium with the underlined chorion or lamina propria. An orthokeratinized epithelium, similar to the skin, is present in the gum—the gingival mucosa—and at the level of the hard palate [42,43,44][42][43][44]. In the basal and intermediate layers of the epithelium, cells involved in immunity may be present through the Kcs, but these are numerous in the chorion. The oral epithelium from the jugal and labial mucosa and the soft palate is non-keratinized [42,45,46][42][45][46]. HPVs are exclusively intraepithelial pathogens, and an epithelial wound creating a discontinuity through the Kcs is necessary in order to allow the access of virions to the basal membrane and the profound cell layer [29,47][29][47]. Since the basal Kcs of the stratified squamous epithelium are the only ones able to divide, they are the initial target of HPV infection. For low-risk HPVs, which do not stimulate keratinocyte proliferation, this is a good hypothesis, but for high-risk HPVs, which interfere with cell proliferation, it is less clear. For a better understanding of high- and low-risk HPV infection and evolution, see [2]. A study on a mouse model highlighted, surprisingly, that HPVs do not initially bind the Kcs in vivo, but first the heparan sulfate proteoglycans (HSPGs) of BM exposed after the upper epithelial trauma via the L1 major capsid protein. HSPG is considered the first receptor. It also emphasizes the natural dependence of some viruses, including HPVs, on HSPGs used as hijacked receptors in order to bind to host cells. This typical interaction occurs through electrostatic interactions between the cell membrane and the virus. After binding to HSPGs, the mature virus particle undergoes a series of conformational changes to enter the host cell [48,49][48][49]. Following these conformational changes, viral capsids can be transferred to a non-HSPG epithelial cell receptor. To date, after binding HSPGs, HPV-16 interacts with the EGFRs present on the plasma membrane and produces an important reconfiguration of the host cells [49]. Once internalized by a micropinocytosis-like mechanism involving actin, the virus traffics through the endolysosomal system and reaches the nucleus [1]. Different viral genes are expressed during the cell-hosting virus superficialization [29], and the expression of viral proteins seems to be regulated by the differentiation of Kcs. Virus multiplication takes place in the internal layers of the epithelium driven by E6-E7 genes, key regulators of cell cycle progression, but genome amplification with viral protein production is noted in the upper epithelial layers, stratum spinosum and stratum granulosum, under the E4 gene control [2,50][2][50]. Here, the number of DNA copies is highly increased, up to a thousand/cell [47], and finally, new virions are packed and leave the epithelium under the control of early E4 and late L1 and L2 genes [1,29][1][29]. The accomplishment of a virus cycle takes time even in optimal conditions, and depending on the oral topographic area, the delay from the infection to the virion release is about 20 days, the time for keratinocyte daughters to reach the stratum corneous from the basal membrane [50]. The pathogenesis of low- and high-risk HPVs is notably different, even in the presence of the same gene products: low-risk HPVs produce a rapid progeny and a large lesion of the mucosa and induce the local immune response and inflammation; E6 and E7 proteins display low activity of transforming the host genome and lack the capacity to produce genomic instability [47]. Depending on the high-risk or low-risk virus genotype, the host immune response, the epigenetic conditions and the topographic region of the oral epithelium, the virus could get cleared, could remain latent or self-servingly drive the cell cycle to replicate and onset the pathological changes.5. HPV Clearance
Clearance is a feature of the majority of HPVs. Usually, 63% to 70% of viral infections cleared rapidly within 12 months in response to an efficient immune system [51]. For most subjects, HPV-associated lesions are cleared within 1–2 years [52]. Virus clearing is associated with sexual behavior, while no epidemiologic link between the oncogenic or non-oncogenic types of viruses and age was noted. The risk of HPV infection decreases with age and is probably linked to a higher presence of HPV antibodies [53]. The median time for clearance of any type of HPV infection was significantly longer in young men as compared to other age groups [40]. Other studies state that the median time of HPV-16 clearance is nearly two times longer than with other oncogenic HPV types [54]. The resolution of infection requires the cross-priming of dendritic cells (DCs) followed by T-cell infiltration into the site of infection and the shut-off of viral gene expression [29]. An interesting study of the imbalance between the effectiveness of the immune system versus the stochastic dynamics of basal stem cells suggests that the chance of the division of basal Kcs plays a critical role in the elimination of HPV-infected cell clones. In subjects with a normal immune capacity, the immune response may contribute to less than 20% of the viral clearing, the rest being ensured by the random succession of symmetric and asymmetric stem cell mitosis [55]. Virus clearance could be illusory as lesion regression is not associated with massive apoptosis or cell death. The lesion is cleared by the replacement of actively infected cells with ‘apparently normal’ cells as the basal cells continue to divide. These ’apparently normal’ cells may still contain viral genomes but without concomitant viral gene expression, and, in animal models, it has been suggested that the virus life cycle may become ‘reactivated’ depending on individual conditions [29].6. HPV Persistence
It is a general agreement that the Kcs of the oral mucosa are the reservoir of HPV, and the most common behavior of the oncogenic HPVs, especially HPV-16 and 18, but also of the low-risk HPV-6 and 11, is the virus persistence [56]. In order to maintain itself inside the host cells, either viral DNA is integrated into the host genome as for high-risk HPVs, or it is maintained as episomes that tether to host DNA as for low-risk HPVs. The integration of HPV DNA alters the expression of key genes in the host and promotes genomic instability and oncogenic progression [56]. HPV-6 and 11 are responsible for RRP that rarely can convert to cancer [36]. It has been reported that this rare oncogenesis of RRP may be caused by the integration of the HPV gene into the host cell genome, thereby knocking out the host gene [57]. Research data provide evidence that HPV-11 poses a higher potential for malignant transformation [57]. The oncogenicity differences between these low-risk HPVs could be related to the ability of E6 and E7 proteins of HPV-11 to have a more efficient interaction with host cell “oncogenic targets”, than those from HPV-6 [36,58][36][58]. It demonstrated the involvement of E2 protein from high-risk HPV-16/18 and HPV-11 to maintain HPV DNA as “mini-chromosomes” in dividing cells through its direct interaction with the mitotic spindles [56]. Still, HPV-6 and 11 proteins have a different ability to interfere with tumor suppressors p53 and pRb and other cell cycle regulators than those from high-risk HPVs [36]. Among the risk factors for virus persistence, there are the immunosuppression of HIV-infected patients and renal transplant recipients and smoking [1], albeit recent studies have described smoking and HPV infection as independent risk factors for oropharyngeal cancer. Subjects with some gene polymorphisms of the major histocompatibility complex (MHC) are also more susceptible to HPV persistence and cancerization, as reviewed in [56]. Research data is not congruent that co-infection with various HPV types is a potential predictor of subsequent persistence of the HPVs [59]. Chronologically, virus persistence is related to the storage of the viral genome in the epithelial stem cells followed by virus reactivation at the site of the previous infection or in other regions [60]. In the nuclei of the Kcs from the epithelial basal layer, the viral genome is maintained as an episome or plasmid (a low copy number), meaning that viral gene expression is minimal [61]. The state of viral genome integration in the host cells, with no viral DNA replication but production of the oncoviral proteins and nuclear mutagenesis, is called “pseudolatency”, three oncogenic genes being expressed in this stage, E5, E6 and E7. E5 gene product stimulates EGF-mediated cell proliferation and affects apoptosis [62,63][62][63]. The E5 protein acts as a viroporin, affecting vesicle trafficking and cellular homeostasis, altering antigen presentation and inflammation [64]. This latent phase can persist for years [1]. In this phase, unlike the low-risk HPVs DNA, the oncogenic high-risk HPVs DNA is integrated into the Kc genome under the action of E6 and E7 that escaped the E2 protein control [47]. This is considered a canonic viral gene imbalance explaining the persistence and carcinogenesis of HPV [65]. The E6 and E7 proteins from low-risk HPVs shared many of the cellular effects of their counterparts from high-risk HPVs but to a generally lesser extent. An explanation for their different oncogenic potential could be related to the different efficiency of action on cellular targets. The research data showed that low-risk HPVs producing RRP might undergo mutations which lead to increased levels of E6 and E7. This could explain why, once penetrated into the lung tissue, these pathogens determine lung cancer [58]. Some events (epigenetic factors such as local irritation) may determine the virus from the proliferative zone of the epithelium to leave the stable replication to their entry into the vegetative mechanism [1], and, as the virus multiplies, to migrate with the Kcs through the epithelium surface [29,47][29][47]. This explains the fact that the oral mucosa, together with the ductal epithelium of the salivary gland, the epithelium of the tonsillar crypts and the epithelium of the gingival pockets may represent an infection reservoir for the remote carcinogenesis of tonsils and pharynx [1,39][1][39]. The production of the HPV-16 early E5 gene ensures the persistence of infection in immortalized HPV-infected Kcs (Kcs) by downregulating the expression of MHC-I molecules through retention in the Golgi apparatus membranes and the inhibition of their transport to the cell surface [66], reducing the susceptibility of Kcs to be cleared by the cytotoxic T lymphocytes (CTL) and Natural Killer (NK) cells [67]. It should be noted that, in contrast to cervix carcinoma and epidermis, in the oral mucosa, virus multiplication is achieved without the damage of oral Kcs, and consequently premalignancies associated with OSCC are unknown [65]. However, a reduced functional impairment of Kcs in advanced stages of oral papillomatosis may occur [68,69][68][69]. It is not evident that a particular area of the epithelium is more susceptible to becoming a transitional host or a long-term reservoir for HPVs. A surprising finding is that among the HNSCN etiology, HPV’s prevalence in the oropharyngeal location is significantly higher in palatines tonsils (61, 8%) and the base of the tongue/lingual tonsil (49, 45%) [1], both areas included in the Waldayer‘s lymphatic ring. It seems that in this location, HPV infection may occur even in the absence of an anterior epithelial wound due to the particular structure of the epithelium, called improperly reticulated epithelium, as its cells and the underline basal membrane are discontinuous. It was speculated that the evolution of an HPV infection into a squamous cancer may be caused by the intimate relationship between epithelial and immune cells [70]. A second interesting site, with a particular histological organization and a huge involvement in the gingival homeostasis, able to host a latent HPV, is the epithelium forming the wall of the gingival pocket. As in the tonsillar criptae, the Kcs dynamically multiply with the faster turnover of the oral cavity, 4–6 days, loose packing and the numerous intercellular spaces created filling with crevicular fluid [71]. The access of HPV through the BM is facilitated and its integration into the genome may be more efficient. The coexistence of epithelial and immune cells is a feature of both locations. Underpinning this idea is the observation that oncogenic HPVs were found concealed in the junctional epithelium of the dental pocket in 26% of the analyzed cases of periodontitis. Whether HPV is a trigger of the periodontal disease or is installed in the slipstream of other pathogens enhancing the local inflammation is to be studied [72,73][72][73]. Epidemiologically, the gum is the most common site of OSSC HPV positive [8].References
- Syrjänen, S. Oral manifestations of human papillomavirus infections. Eur. J. Oral Sci. 2018, 126 (Suppl. S1), 49–66.
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23.
- Lisan, Q.; Laccourreye, O.; Bonfils, P. Sinonasal inverted papilloma: From diagnosis to treatment. Eur. Ann. Otorhinolaryngol. Head Neck. Dis. 2016, 133, 337–341.
- Sabry, A.O.; Patel, B.C. Papilloma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560737/ (accessed on 17 June 2022).
- Gupta, I.; Jabeen, A.; Al-Sarraf, R.; Farghaly, H.; Vranic, S.; Sultan, A.A.; Al Moustafa, A.-E.; Al-Thawadi, H. The co-presence of high-risk human papillomaviruses and Epstein-Barr virus is linked with tumor grade and stage in Qatari women with breast cancer. Hum. Vaccin. Immunother. 2021, 17, 982–989.
- Marshall, J.R.; Graham, S.; Haughey, B.P.; Shedd, D.; O’Shea, R.; Brasure, J.; Wilkinson, G.S.; West, D. Smoking, alcohol, dentition and diet in the epidemiology of oral cancer. Eur. J. Cancer B Oral Oncol. 1992, 28, 9–15.
- Behnoud, F.; Torabian, S.; Zargaran, M. Relationship between oral poor hygiene and broken teeth with oral tongue squamous cell carcinoma. Acta Med. Iran. 2011, 49, 159–162.
- Gupta, S.; Gupta, S. Role of human papillomavirus in oral squamous cell carcinoma and oral potentially malignant disorders: A review of the literature. Indian J. Dent. 2015, 6, 91–98.
- Ma, J.; Zhang, J.; Zhang, Y.; Lv, T.; Liu, J. The magnitude of the association between human papillomavirus and oral lichen planus: A meta-analysis. PLoS ONE 2016, 11, e0161339.
- Razavi, S.M.; Ghalayani, P.; Salehi, M.R.; Attarzadeh, H.; Shahmoradi, M. Human papilloma virus as a possible factor in the pathogenesis of oral lichen planus. Dent. Res. J. 2009, 6, 82–86.
- Villa, T.G.; Sánchez-Pérez, Á.; Sieiro, C. Oral lichen planus: A microbiologist point of view. Int. Microbiol. 2021, 24, 275–289.
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92.
- Sun, Z.; Sun, X.; Chen, Z.; Du, J.; Wu, Y. Head and Neck Squamous Cell Carcinoma: Risk Factors, Molecular Alterations, Immunology and Peptide Vaccines. Int. J. Pept. Res. Ther. 2022, 28, 19.
- Faraji, F.; Zaidi, M.; Fakhry, C.; Gaykalova, D.A. Molecular mechanisms of human papillomavirus-related carcinogenesis in head and neck cancer. Microbes Infect. 2017, 19, 464–475.
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72.
- Zaravinos, A. An updated overview of HPV-associated head and neck carcinomas. Oncotarget 2014, 5, 3956–3969.
- Camuzi, D.; Simão, T.A.; Dias, F.; Ribeiro Pinto, L.F.; Soares-Lima, S.C. Head and Neck Cancers Are Not Alike when Tarred with the Same Brush: An Epigenetic Perspective from the Cancerization Field to Prognosis. Cancers 2021, 13, 5630.
- Marur, S.; Forastiere, A.A. Head and neck cancer: Changing epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 2008, 83, 489–501.
- Lechien, J.R.; Descamps, G.; Seminerio, I.; Furgiuele, S.; Dequanter, D.; Mouawad, F.; Badoual, C.; Journe, F.; Saussez, S. HPV Involvement in the Tumor Microenvironment and Immune Treatment in Head and Neck Squamous Cell Carcinomas. Cancers 2020, 12, 1060.
- Shrestha, A.D.; Neupane, D.; Vedsted, P.; Kallestrup, P. Cervical Cancer Prevalence, Incidence and Mortality in Low and Middle Income Countries: A Systematic Review. Asian Pac. J. Cancer Prev. 2018, 19, 319–324.
- Taberna, M.; Mena, M.; Pavón, M.A.; Alemany, L.; Gillison, M.L.; Mesía, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398.
- Bednarczyk, R.A. Addressing HPV vaccine myths: Practical information for healthcare providers. Hum. Vaccin. Immunother. 2019, 15, 1628–1638.
- Hirth, J. Disparities in HPV vaccination rates and HPV prevalence in the United States: A review of the literature. Hum. Vaccin. Immunother. 2019, 15, 146–155.
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32.
- Kumaraswamy, K.L.; Vidhya, M. Human papilloma virus and oral infections: An update. J. Cancer Res. Ther. 2011, 7, 120–127.
- Fiorillo, L.; Cervino, G.; Surace, G.; De Stefano, R.; Laino, L.; D’Amico, C.; Fiorillo, M.T.; Meto, A.; Herford, A.S.; Arzukanyan, A.V.; et al. Human Papilloma Virus: Current Knowledge and Focus on Oral Health. BioMed Res. Int. 2021, 2021, 6631757.
- Wang, H.F.; Wang, S.S.; Tang, Y.J.; Chen, Y.; Zheng, M.; Tang, Y.L.; Liang, X.H. The Double-Edged Sword-How Human Papillomaviruses Interact With Immunity in Head and Neck Cancer. Front. Immunol. 2019, 10, 653.
- Du, J.; Ährlund-Richter, A.; Näsman, A.; Dalianis, T. Human papilloma virus (HPV) prevalence upon HPV vaccination in Swedish youth: A review based on our findings 2008–2018, and perspectives on cancer prevention. Arch. Gynecol. Obstet. 2021, 303, 329–335.
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. S5), F55–F70.
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27.
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322.
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475.
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22.
- Melo, B.A.C.; Vilar, L.G.; Oliveira, N.R.; Lima, P.O.; Pinheiro, M.B.; Domingueti, C.P.; Pereira, M.C. Human papillomavirus infection and oral squamous cell carcinoma—A systematic review. Rev. Bras. Otorrinolaringol. 2021, 87, 346–352.
- McCord, C.; Xu, J.; Xu, W.; Qiu, X.; Muhanna, N.; Irish, J.; Leong, I.; McComb, R.J.; Perez-Ordonez, B.; Bradley, G. Association of human papilloma virus with atypical and malignant oral papillary lesions. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 722–732.
- Ivancic, R.; Iqbal, H.; de Silva, B.; Pan, Q.; Matrka, L. Immunological tolerance of low-risk HPV in recurrent respiratory papillomatosis. Clin. Exp. Immunol. 2020, 199, 131–142.
- Sampaio, J.; Ferreira, J.; Santos, A.C.; Bicho, M.; Bicho, M.C. The Importance of the Extracellular Matrix in HPV-Associated Diseases. In Cervical Cancer—A Global Public Health Treatise; Rajkumar, R., Ed.; IntechOpen: London, UK, 2021; Available online: https://www.intechopen.com/chapters/78569 (accessed on 17 June 2022).
- Münger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228.
- Candotto, V.; Lauritano, D.; Nardone, M.; Baggi, L.; Arcuri, C.; Gatto, R.; Gaudio, R.M.; Spadari, F.; Carinci, F. HPV infection in the oral cavity: Epidemiology, clinical manifestations and relationship with oral cancer. Oral Implantol. 2017, 10, 209–220.
- Bharti, A.H.; Chotaliya, K.; Marfatia, Y.S. An update on oral human papillomavirus infection. Indian J. Sex. Transm. Dis. AIDS 2013, 34, 77–82.
- Castro, T.P.; Bussoloti Filho, I. Prevalence of human papillomavirus (HPV) in oral cavity and oropharynx. Rev. Bras. Otorrinolaringol. 2006, 72, 272–282.
- Winning, T.A.; Townsend, G.C. Oral mucosal embryology and histology. Clin. Dermatol. 2000, 18, 499–511.
- Brizuela, M.; Winters, R. Histology, Oral Mucosa. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK572115/ (accessed on 17 June 2022).
- Nikoloudaki, G.; Creber, K.; Hamilton, D.W. Wound healing and fibrosis: A contrasting role for periostin in skin and the oral mucosa. Am. J. Physiol. Cell Physiol. 2020, 318, C1065–C1077.
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208.
- Otsuka-Tanaka, Y.; Oommen, S.; Kawasaki, M.; Kawasaki, K.; Imam, N.; Jalani-Ghazani, F.; Hindges, R.; Sharpe, P.T.; Ohazama, A. Oral lining mucosa development depends on mesenchymal microRNAs. J. Dent. Res. 2013, 92, 229–234.
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus immune evasion strategies target the infected cell and the local immune system. Front. Oncol. 2019, 9, 682.
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463.
- Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19, 1775.
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222.
- Rodríguez, A.C.; Schiffman, M.; Herrero, R.; Wacholder, S.; Hildesheim, A.; Castle, P.E.; Solomon, D.; Burk, R.; Proyecto Epidemiológico Guanacaste Group. Rapid clearance of human papillomavirus and implications for clinical focus on persistent infections. J. Natl. Cancer Inst. 2008, 100, 513–517.
- Mishra, G.A.; Pimple, S.A.; Shastri, S.S. An overview of prevention and early detection of cervical cancers. Indian J. Med. Paediatr. Oncol. 2011, 32, 125–132.
- Markowitz, L.E.; Schiller, J.T. Human Papillomavirus Vaccines. J. Infect. Dis. 2021, 224 (Suppl. S2), S367–S378.
- Giuliano, A.R.; Lee, J.H.; Fulp, W.; Villa, L.L.; Lazcano, E.; Papenfuss, M.R.; Abrahamsen, M.; Salmeron, J.; Anic, G.M.; Rollison, D.E.; et al. Incidence and clearance of genital human papillomavirus infection in men (HIM): A cohort study. Lancet 2011, 377, 932–940.
- Ryser, M.D.; Myers, E.R.; Durrett, R. HPV clearance and the neglected role of stochasticity. PLoS Comput. Biol. 2015, 11, e1004113.
- Shanmugasundaram, S.; You, J. Targeting Persistent Human Papillomavirus Infection. Viruses 2017, 9, 229.
- Huebbers, C.U.; Preuss, S.F.; Kolligs, J.; Vent, J.; Stenner, M.; Wieland, U.; Silling, S.; Drebber, U.; Speel, E.J.; Klussmann, J.P. Integration of HPV6 and downregulation of AKR1C3 expression mark malignant transformation in a patient with juvenile-onset laryngeal papillomatosis. PLoS ONE 2013, 8, e57207.
- Pim, D.; Banks, L. Interaction of viral oncoproteins with cellular target molecules: Infection with high-risk vs. low-risk human papillomaviruses. Apmis 2010, 118, 471–493.
- Liaw, K.L.; Hildesheim, A.; Burk, R.D.; Gravitt, P.; Wacholder, S.; Manos, M.M.; Scott, D.R.; Sherman, M.E.; Kurman, R.J.; Glass, A.G.; et al. A prospective study of human papillomavirus (HPV) type 16 DNA detection by polymerase chain reaction and its association with acquisition and persistence of other HPV types. J. Infect. Dis. 2001, 183, 8–15.
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163.
- Myers, J.E.; Guidry, J.T.; Scott, M.L.; Zwolinska, K.; Raikhy, G.; Prasai, K.; Bienkowska-Haba, M.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Detecting episomal or integrated human papillomavirus 16 DNA using an exonuclease V-qPCR-based assay. Virology 2019, 537, 149–156.
- Vescovo, T.; Pagni, B.; Piacentini, M.; Fimia, G.M.; Antonioli, M. Regulation of Autophagy in Cells Infected with Oncogenic Human Viruses and Its Impact on Cancer Development. Front. Cell Dev. Biol. 2020, 8, 47.
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human oncogenic viruses: Nature and discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160264.
- Mikuličić, S.; Florin, L. The endocytic trafficking pathway of oncogenic papillomaviruses. Papillomavirus Res. 2019, 7, 135–137.
- Wittekindt, C.; Wagner, S.; Sharma, S.J.; Würdemann, N.; Knuth, J.; Reder, H.; Klußmann, J.P. HPV—A different view on Head and Neck Cancer. HPV—Das andere Kopf-Hals-Karzinom. Laryngo-Rhino-Otologie 2018, 97 (Suppl. S1), S48.
- Ashrafi, G.H.; Brown, D.R.; Fife, K.H.; Campo, M.S. Down-regulation of MHC class I is a property common to papillomavirus E5 proteins. Virus Res. 2006, 120, 208–211.
- Cortese, M.S.; Ashrafi, G.H.; Campo, M.S. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 2010, 126, 1675–1682.
- Franceschi, S.; Muñoz, N.; Bosch, X.F.; Snijders, P.J.; Walboomers, J.M. Human papillomavirus and cancers of the upper aerodigestive tract: A review of epidemiological and experimental evidence. Cancer Epidemiol. Biomarkers Prev. 1996, 5, 567–575.
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980.
- Paz, I.B.; Cook, N.; Odom-Maryon, T.; Xie, Y.; Wilczynski, S.P. Human papillomavirus (HPV) in head and neck cancer. An association of HPV 16 with squamous cell carcinoma of Waldeyer’s tonsillar ring. Cancer 1997, 79, 595–604.
- Fehrenbach, M.J.; Popowics, T. Illustrated Dental Embryology, Histology, and Anatomy, 4th ed.; Elssevier Saunders: Maryland Heights, MO, USA, 2016; pp. 9–17.
- Hormia, M.; Willberg, J.; Ruokonen, H.; Syrjänen, S. Marginal periodontium as a potential reservoir of human papillomavirus in oral mucosa. J. Periodontol. 2005, 76, 358–363.
- Tezal, M.; Sullivan Nasca, M.; Stoler, D.L.; Melendy, T.; Hyland, A.; Smaldino, P.J.; Rigual, N.R.; Loree, T.R. Chronic periodontitis-human papillomavirus synergy in base of tongue cancers. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 391–396.