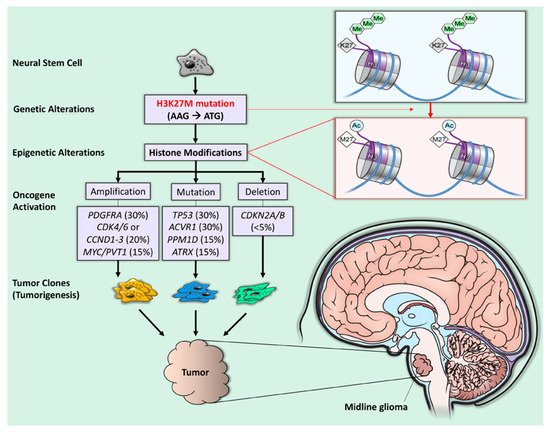
4.3. Targeting ACVR1 Mutation
Although most preclinical studies of diffuse midline glioma target H3K27M-related mechanisms,
ACVR1 mutations remain an important driver of tumorigenesis in more than 30% of H3K27M-mutant diffuse midline gliomas
[31][34][40,48]. Since ACVR1 encodes the serine/threonine kinase (ALK2), ALK2 inhibitors could play a role in H3K27M DMG treatment. Recently, ALK2 inhibition improved survival in orthotopic xenograft mice bearing H3.3K27M, ACVR1R206H tumors compared to control mice without the ACVR mutation
[35][49].
4.4. EZH2 Inhibition
Overexpression of enhancer of zeste homolog 2 (EZH2), an enzyme involved in histone methylation, has been associated with a more dismal prognosis in DMG patients with H3K27M mutations
[36][52]. Mohammad et al. found that small-molecule EZH2 inhibitors abolished tumor growth in an H3K27M-mutant diffuse midline glioma mouse model by inducing protein p16
INK4A, a tumor-suppressing molecule
[37][53].
4.5. Metabolic Inhibitors
Several other approaches for targeting H3K27M DMG have also been proposed that target cellular metabolic pathways. Recently, Khan et al. demonstrated that polyamine synthesis is upregulated in DIPG and, therefore, could be targeted through a synthetic lethality-based approach using a polyamine synthesis inhibitor, difluoromethylornithine (DFMO). Since DIPG cells preferentially escape DFMO inhibition through upregulation of the polyamine transporter SLC3A2, adding polyamine transport inhibitors (AMXT 1501) potentiated tumor-selective cytotoxicity in vitro and in orthotopic animal models
[38][55].
4.6. Immunotherapy
Several studies have investigated immunotherapy as a potential therapy for H3K27M DMG. Preclinical research for H3K27M DMG has incorporated recent chimeric antigen receptor (CAR) T-cell therapy advances. CAR T-cell therapy directed against GD2 (disialoganglioside), a tumor-associated cell surface antigen, is intriguing because patient-derived H3K27M DMG cell lines and neuroectodermal tissues almost uniformly express GD2
[39][57]. Additionally, in vitro exposure to anti-GD2-CAR T cells significantly depleted cultured H3K27M-mutant cells in a dose-dependent manner
[39][57]. However, the brain penetrance of this CAR T-cell directed therapy is uncertain. Mount et al. did demonstrate brain penetrance in their murine xenograft model, although such penetrance was associated with neurotoxicities including peritumoral inflammation and resultant hydrocephalus
[39][57]. Nevertheless, the tumoricidal effect of GD2-targeted CAR T cells in in vitro assays supports further development and testing of this approach in animal models. Interestingly, immunotherapeutic trials for neuroblastoma, osteosarcoma, and melanoma which target GD2 were successful
[39][57].
54. Management
5.1. Radiation Therapy
4.2. Radiation Therapy
Radiation therapy is the only treatment that improves life expectancy. A 54–60 Gy radiotherapy tumor dose over 6 weeks has been the standard treatment recommendation for H3K27M-mutant diffuse midline glioma over the past 20 years, delaying tumor progression for up to 3 months in 70–80% of patients
[2][40][41][2,62,63]. Hyperfractionated therapy is less effective than conventional therapy and may increase radiation toxicity and other morbidities
[42][43][44][64,65,66]. However, hypofractionated radiotherapy may reduce the burden of treatment for caretakers by significantly reducing hospitalizations and improving the quality of life
[45][46][67,68].
Radiation may also have synergistic effects if combined with chemotherapy or immunotherapy for treating H3K27M-mutant DMG. Radiosensitizing agents such as gemcitabine selectively target rapidly dividing tumor cells and potentiate radiotherapy. In a phase I/II study, gemcitabine, a pyrimidine analog, and concurrent radiotherapy were well tolerated and without dose-limiting toxicity. However, overall survival was similar to that of historical controls (mOS of 8.7 months)
[47][69].
5.2. Tumor-Localized Therapy
4.3. Tumor-Localized Therapy
Technological and neurosurgical advancements in stereotactic MRI-guided infusion catheter placement, catheter design, and drug distribution monitoring have paved the way for clinical studies and trials of intratumoral drug delivery via convection-enhanced delivery (CED)
[48][75]. CED is a regional drug delivery method utilizing small hydrostatic pressure gradients to drive the bulk flow of a drug through the extracellular spaces of the central nervous system. CED is a promising therapeutic delivery technique for treating patients with H3K27M-mutant diffuse midline glioma. Several earlier studies have established the technical parameters for safely using CED in patients with H3K27M-mutant DMG
[49][50][51][52][76,77,78,79].
Two clinical trials using CED to treat H3K27M-mutant diffuse midline glioma have recently been reported. Heiss et al. investigated the safety, infusion distribution, and potential efficacy of CED of IL13–
Pseudomonas exotoxin in five pediatric patients with H3K27M-mutant diffuse midline glioma
[53][10]. They observed short-term radiographic antitumor effects in two of the five patients treated. However, the IL13–
Pseudomonas exotoxin was not distributed widely enough to reach the entire MRI-defined tumor volume in any patient. Poor target drug distribution contributed to the lack of efficacy in this trial. Drug distribution could have been improved by infusing through multiple catheters or priming the tumor microenvironment. In another clinical trial, Souweidane et al. used CED to safely infuse a murine monoclonal antibody targeting glioma-associated B7-H3 antigen, conjugated to the radioisotope conjugate ([
124I]-8H9) into the brainstem of pediatric patients with H3K27M-mutant DMG
[54][72]. The primary endpoint was the maximum tolerated dose of [
124I]-8H9 administered by convection-enhanced delivery. No dose-limiting toxicities occurred with any dose, and the maximum-tolerated dose was not reached. Overall survival in this group was slightly better than historical controls (15.3 months). Survival increased more in patients in higher-dose cohorts.
5.3. Immunotherapy
4.4. Immunotherapy
Immunotherapy of H3K27M-mutant diffuse midline glioma is being tested as an adjuvant to traditional chemoradiation. Immunotherapy was safe when administered intravenously and concomitantly with radiotherapy
[54][55][56][71,72,74]. Benitez-Ribas et al. recently reported a phase Ib immunotherapy clinical trial using autologous dendritic cells pulsed with an allogeneic tumor cell-line lysate to reactivate tumor-specific T cells in patients with newly diagnosed H3K27M-mutant diffuse midline glioma after irradiation
[57][88]. Autologous dendritic cell vaccines were feasible, safely prepared, and generated an H3K27M-mutant diffuse midline glioma-specific immune response detected in peripheral blood mononuclear cells (PBMC) and cerebrospinal fluid (CSF). In another study, Fried et al. demonstrated that the immune-modulating antibody MDV9300 (pidilizumab) is a potentially promising treatment for H3K27M-mutant DMG following radiotherapy
[58][89]. Of the nine pediatric patients enrolled in the study, two were still alive nearly 30 months from diagnosis at the trial’s conclusion, with radiographically defined disease stability
[4][58][4,89]. Additionally, Tejada et al. reported the novel intratumoral use of DNX-2401, a replication-competent, genetically modified virus that stimulates an antitumor immune response, in a pediatric patient with H3K27M-mutant diffuse midline glioma
[59][22]. Preclinical studies suggested that DNX-2401 (known as Delta-24-RGD) effectively induced both an oncolytic and an antitumor immune response in pediatric high-grade gliomas
[60][90]. DNX-2401 is under investigation in an ongoing phase I clinical trial (NCT03178032) for recently diagnosed H3K27M-mutant DMG patients. Their findings further suggest that immunotherapy is a promising adjunctive treatment for H3K27M-mutant diffuse midline glioma.