Recent data suggests that the onset of prediabetes and diabetes are preceded by a variable period of hyperinsulinemia. Emerging data suggest that chromic hyperinsulinemia is also a driving force for increased activation of the hypothalamic-adrenal-pituitary (HPA) axis in subjects with the metabolic syndrome, leading to a state of “functional hypercortisolism”. This “functional hypercortisolism” by antagonizing insulin actions may prevent hypoglycemia. It also disturbs energy balance by shifting energy fluxes away from muscles toward abdominal fat stores. Synergistic effects of hyperinsulinemia and “functional hypercortisolism” promote abdominal visceral obesity and insulin resistance which are core pathophysiological components of the metabolic syndrome. It is hypothesized that hyperinsulinemia-induced increased activation of the HPA axis plays an important etiological role in the development of the metabolic syndrome and its consequences. Numerous studies have demonstrated reversibility of hyperinsulinemia with lifestyle, surgical, and pharmaceutical-based therapies. Longitudinal studies should be performed to investigate whether strategies that reduce hyperinsulinemia at an early stage are successfully in preventing increased activation of the HPA axis and the metabolic syndrome.
- hyperinsulinemia
- relative hypoinsulinemia
- insulin
- insulin resistance
- insulin receptor
- westernized diet
- over-nutition
- metabolic syndrome
- Cushing syndrome
- cortisoluria
1. Introduction


| Metabolic Syndrome | Cushing Syndrome |
|
|---|---|---|
| Plasma Insulin levels | ↑↑ | ↑ * or ↓ ** |
| insulin resistance | + | + |
| AbdominalObesity | + | + |
| impaired glucose tolerance | + | + |
| Hypertriglyceridemia | + | + |
| Hypertension | + | + |
| HPA Activity | ↑ | ↑ |
| Plasma Cortisol | ↓/N | ↑ |
| 24 h-Free Cortisoluria | ↑ | ↑ |
2. Regulation of the Hypothalamic-Pituitary-Adrenal (HPA)-Axis in Healthy Subjects
Hypothalamus and pituitary regulate cortisol synthesis and release in healthy subjects (Figure 3). Corticotropin-releasing hormone (CRH) is released by the hypothalamus and stimulates the anterior pituitary to release ACTH. ACTH then acts on the adrenal cortex which promotes the secretion of cortisol from the zona fasciculata. The secretion of cortisol provides a negative feedback loop by inhibiting release of CRH and ACTH from the hypothalamus and anterior pituitary, respectively. The activity of 11 beta-hydroxysteroid dehydrogenase (11b-HSD) plays an important role in extra-adrenal cortisol metabolism [24]. At least two isozymes of 11 beta-HSD exist, which catalyze the interconversion of hormonally active glucocorticoids (cortisol, corticosterone) and their inactive metabolites (cortisone, 11-dehydrocorticosterone) [25] 11β-hydroxysteroid dehydrogenase-2 (11β-HSD2), is mainly expressed in the kidneys and protects the mineralocorticoid receptor from glucocorticoid excess by converting cortisol to cortisone [25]. It thereby promotes the access of aldosterone to the mineralocorticoid receptors in the kidney. The other isoform, 11β-hydroxysteroid dehydrogenase-1 (11β-HSD1), is widely expressed in classic insulin target tissues as the liver, muscle, and adipose tissue.
3. The Bidirectional Interactions between Insulin and the HPA Axis
In healthy subjects, insulin normally shows a reciprocal relationship with cortisol: insulin inhibits food intake while cortisol stimulates food intake [31]. Insulin and cortisol are major antagonistic regulators of energy balance. Effects of cortisol and insulin on food intake may be mediated through regulation of hypothalamic neuropeptide-Y (NPY) synthesis and secretion [31]. In the arcuate nuclei, insulin inhibits and cortisol stimulates the expression of NPY mRNA, which may explain in part the reciprocal actions of insulin and the HPA axis on energy acquisition during the day. It has been further suggested that inhibition of insulin transport across the blood brain barrier by glucocorticoids could be the basis for the enhanced appetite seen with glucocorticoid treatments [32].Chronic exposure to high circulating glucocorticoid levels inhibits insulin release by binding to glucocorticoid receptors present on pancreatic beta cells [23].Chronic exposure to glucocorticoids may further reduce the insulinotropic effects of glucagon-like peptide-1 (GLP-1) [33]. Thus, with several mechanisms, glucocorticoids may inhibit pancreatic β-cell mediated insulin secretion and thereby induce a “relative hypoinsulinemia”. In addition, glucocorticoids may induce insulin resistance (see next paragraph Glucocorticoids, insulin resistance and metabolism). As long as pancreatic insulin secretion is sufficient, cortisol-mediated increase of insulin resistance and hepatic glucose production will not materially affect glucose tolerance. However, failure of the pancreas to mount an adequate compensatory insulinemic response (due to cortisol-mediated suppression of pancreatic insulin release) may lead to hyperglycemia and impaired glucose tolerance (Figure 4).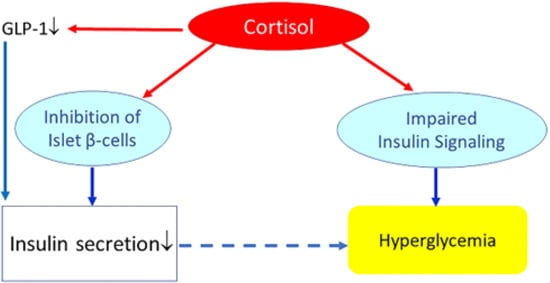 Figure 4. Effects of (high) glucocorticoids on insulin, insulin resistance, and glucose metabolism. Cortisol directly suppresses pancreatic insulin release. It also reduces glucagon-like peptide-1 (GLP-1) production, which further decreases insulin secretion. Increased cortisol also induces glycogenolysis and the expression of key gluconeogenic enzymes, which will increase hepatic glucose production and release. In addition, cortisol may impair insulin receptor signaling (at the receptor and post-receptor level) and thereby induce insulin resistance. Failure of the pancreas to mount an adequate compensatory insulinemic response (“relative hypoinsulinemia” due to cortisol-mediated suppression of pancreatic insulin release) with cortisol-induced insulin resistance may lead to hyperglycemia and impaired glucose tolerance. However, if insulin secretion is sufficient to overcome cortisol-mediated insulin resistance, cortisol will not materially affect glucose tolerance.
Figure 4. Effects of (high) glucocorticoids on insulin, insulin resistance, and glucose metabolism. Cortisol directly suppresses pancreatic insulin release. It also reduces glucagon-like peptide-1 (GLP-1) production, which further decreases insulin secretion. Increased cortisol also induces glycogenolysis and the expression of key gluconeogenic enzymes, which will increase hepatic glucose production and release. In addition, cortisol may impair insulin receptor signaling (at the receptor and post-receptor level) and thereby induce insulin resistance. Failure of the pancreas to mount an adequate compensatory insulinemic response (“relative hypoinsulinemia” due to cortisol-mediated suppression of pancreatic insulin release) with cortisol-induced insulin resistance may lead to hyperglycemia and impaired glucose tolerance. However, if insulin secretion is sufficient to overcome cortisol-mediated insulin resistance, cortisol will not materially affect glucose tolerance.
4. Hyperinsulinemia Induces a State of “Functional Hypercortisolism”
As previously discussed, numerous recent data are supportive of the concept that hyperinsulinemia per se is primary and causes insulin resistance [3,11][3][11] (Figure 1). In this alternate concept, insulin resistance is proposed to be a physiological defense mechanism of the body preventing hyperinsulinemia-induced hypoglycemia and protecting against overstimulation of target tissues from metabolic stress and nutrient-induced injury [12,13,14][12][13][14]. This concept is even more interesting against emerging data suggesting that chromic hyperinsulinemia is also a driving force for increased activation of the hypothalamic-adrenal-pituitary (HPA) axis in subjects with the metabolic syndrome, leading to a state of “functional hypercortisolism”. Thus hyperinsulinemia-induced increased activation of the HPA axis plays an important etiological role in the development of the metabolic syndrome and its consequences. At this moment it is unclear whether there is a successful strategy to modify hyperinsulinemia-induced “functional hypercortisolism” in subjects prone to develop the metabolic syndrome.5. Conclusions
In conclusion, emerging data suggest that chromic hyperinsulinemia is the driving force for increased HPA axis activity in subjects with the metabolic syndrome. This “functional hypercortisolism” by antagonizing insulin actions may prevent hypoglycemia, disturb energy homeostasis, and shift energy fluxes away from muscle toward fat stores. Synergistic effects of hyperinsulinemia and “functional hypercortisolism” promote fat accumulation in visceral fat cells and so contribute to abdominal obesity. Chronic hyperinsulinemia-induced activation of the HPA axis may play an important etiological role in the development of the metabolic syndrome and all its consequences.(see J.A.M.J.L. Janssen. New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome, nt. J. Mol. Sci. 2022, 23(15), 8178; https://doi.org/10.3390/ijms23158178 for more details). References.- Reaven, G. Insulin resistance, type 2 diabetes mellitus, and cardiovascular disease: The end of the beginning. Circulation 2005, 112, 3030–3032. [Google Scholar] [CrossRef] [PubMed]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef][Green Version]
- Corkey, B.E. Diabetes: Have we got it all wrong? Insulin hypersecretion and food additives: Cause of obesity and diabetes? Diabetes Care 2012, 35, 2432–2437. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mirzadeh, Z.; Faber, C.L.; Schwartz, M.W. Central Nervous System Control of Glucose Homeostasis: A Therapeutic Target for Type 2 Diabetes? Annu. Rev. Pharmacol. Toxicol. 2022, 62, 55–84. [Google Scholar] [CrossRef] [PubMed]
- Trico, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 2018, 3, e124912. [Google Scholar] [CrossRef][Green Version]
- Brons, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009, 587, 2387–2397. [Google Scholar] [CrossRef]
- Ferrannini, E.; Natali, A.; Bell, P.; Cavallo-Perin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173. [Google Scholar] [CrossRef]
- Loves, S.; van Groningen, L.; Filius, M.; Mekking, M.; Brandon, T.; Tack, C.J.; Hermus, A.; de Boer, H. High-Dose, Diazoxide-Mediated Insulin Suppression Boosts Weight Loss Induced by Lifestyle Intervention. J. Clin. Endocrinol. Metab. 2018, 103, 4014–4022. [Google Scholar] [CrossRef][Green Version]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: A preliminary report. Diabetes Care 2009, 32, 1464–1466. [Google Scholar] [CrossRef][Green Version]
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75. [Google Scholar] [CrossRef]
- Corkey, B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes 2012, 61, 4–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Ruderman, N.B.; Kahn, S.E.; Pedersen, O.; Prentki, M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subsets of type 2 diabetes. Diabetes 2015, 64, 673–686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979. [Google Scholar] [CrossRef]
- Abraham, S.B.; Rubino, D.; Sinaii, N.; Ramsey, S.; Nieman, L.K. Cortisol, obesity, and the metabolic syndrome: A cross-sectional study of obese subjects and review of the literature. Obesity 2013, 21, E105–E117. [Google Scholar] [CrossRef][Green Version]
- Rosmond, R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology 2005, 30, 1–10. [Google Scholar] [CrossRef]
- Anagnostis, P.; Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009, 94, 2692–2701. [Google Scholar] [CrossRef][Green Version]
- Stulnig, T.M.; Waldhausl, W. 11beta-Hydroxysteroid dehydrogenase Type 1 in obesity and Type 2 diabetes. Diabetologia 2004, 47, 1–11. [Google Scholar] [CrossRef][Green Version]
- Duclos, M.; Marquez Pereira, P.; Barat, P.; Gatta, B.; Roger, P. Increased cortisol bioavailability, abdominal obesity, and the metabolic syndrome in obese women. Obes. Res. 2005, 13, 1157–1166. [Google Scholar] [CrossRef]
- Iida, S.; Nakamura, Y.; Fujii, H.; Nishimura, J.; Tsugawa, M.; Gomi, M.; Fukata, J.; Tarui, S.; Moriwaki, K.; Kitani, T. A patient with hypocortisolism and Cushing’s syndrome-like manifestations: Cortisol hyperreactive syndrome. J. Clin. Endocrinol. Metab. 1990, 70, 729–737. [Google Scholar] [CrossRef]
- Friedman, T.C.; Mastorakos, G.; Newman, T.D.; Mullen, N.M.; Horton, E.G.; Costello, R.; Papadopoulos, N.M.; Chrousos, G.P. Carbohydrate and lipid metabolism in endogenous hypercortisolism: Shared features with metabolic syndrome X and NIDDM. Endocr. J. 1996, 43, 645–655. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beaupere, C.; Liboz, A.; Feve, B.; Blondeau, B.; Guillemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623. [Google Scholar] [CrossRef] [PubMed]
- Scaroni, C.; Zilio, M.; Foti, M.; Boscaro, M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr. Rev. 2017, 38, 189–219. [Google Scholar] [CrossRef]
- Traish, A.M.; Guay, A.T.; Zitzmann, M. 5alpha-Reductase inhibitors alter steroid metabolism and may contribute to insulin resistance, diabetes, metabolic syndrome and vascular disease: A medical hypothesis. Horm. Mol. Biol. Clin. Investig. 2014, 20, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Krozowski, Z.S. 11 beta-Hydroxysteroid dehydrogenase. Vitam. Horm. 1999, 57, 249–324. [Google Scholar]
- Tomlinson, J.W.; Stewart, P.M. Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract. Res. Clin. Endocrinol. Metab. 2001, 15, 61–78. [Google Scholar] [CrossRef]
- Napolitano, A.; Voice, M.W.; Edwards, C.R.; Seckl, J.R.; Chapman, K.E. 11Beta-hydroxysteroid dehydrogenase 1 in adipocytes: Expression is differentiation-dependent and hormonally regulated. J. Steroid Biochem. Mol. Biol. 1998, 64, 251–260. [Google Scholar] [CrossRef]
- Janssen, J.; Lamberts, S. Diabetes associated with glucocorticoid excess. In Diabetes Secondary to Endocrine and Pancreatic Disorders; Ghigo, E., Porta, M., Eds.; Karger: Basel, Switzerland, 2014; pp. 22–33. [Google Scholar]
- Dallman, M.F.; Akana, S.F.; Strack, A.M.; Hanson, E.S.; Sebastian, R.J. The neural network that regulates energy balance is responsive to glucocorticoids and insulin and also regulates HPA axis responsivity at a site proximal to CRF neurons. Ann. N. Y. Acad. Sci. 1995, 771, 730–742. [Google Scholar] [CrossRef]
- Goodman, H.M. Basic Medical Endocrinology, 4th ed.; Elsevier: San Diego, CA, USA, 2009. [Google Scholar]
- Strack, A.M.; Sebastian, R.J.; Schwartz, M.W.; Dallman, M.F. Glucocorticoids and insulin: Reciprocal signals for energy balance. Am. J. Physiol. 1995, 268, R142–R149. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Vilsboll, T.; Bagger, J.I.; Holst, J.J.; Knop, F.K. Reduced glucose tolerance and insulin resistance induced by steroid treatment, relative physical inactivity, and high-calorie diet impairs the incretin effect in healthy subjects. J. Clin. Endocrinol. Metab. 2010, 95, 3309–3317. [Google Scholar] [CrossRef][Green Version]
References
- Reaven, G. Insulin resistance, type 2 diabetes mellitus, and cardiovascular disease: The end of the beginning. Circulation 2005, 112, 3030–3032.
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268.
- Corkey, B.E. Diabetes: Have we got it all wrong? Insulin hypersecretion and food additives: Cause of obesity and diabetes? Diabetes Care 2012, 35, 2432–2437.
- Mirzadeh, Z.; Faber, C.L.; Schwartz, M.W. Central Nervous System Control of Glucose Homeostasis: A Therapeutic Target for Type 2 Diabetes? Annu. Rev. Pharmacol. Toxicol. 2022, 62, 55–84.
- Trico, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 2018, 3, e124912.
- Brons, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009, 587, 2387–2397.
- Ferrannini, E.; Natali, A.; Bell, P.; Cavallo-Perin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173.
- Loves, S.; van Groningen, L.; Filius, M.; Mekking, M.; Brandon, T.; Tack, C.J.; Hermus, A.; de Boer, H. High-Dose, Diazoxide-Mediated Insulin Suppression Boosts Weight Loss Induced by Lifestyle Intervention. J. Clin. Endocrinol. Metab. 2018, 103, 4014–4022.
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: A preliminary report. Diabetes Care 2009, 32, 1464–1466.
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75.
- Corkey, B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes 2012, 61, 4–13.
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797.
- Nolan, C.J.; Ruderman, N.B.; Kahn, S.E.; Pedersen, O.; Prentki, M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subsets of type 2 diabetes. Diabetes 2015, 64, 673–686.
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979.
- Abraham, S.B.; Rubino, D.; Sinaii, N.; Ramsey, S.; Nieman, L.K. Cortisol, obesity, and the metabolic syndrome: A cross-sectional study of obese subjects and review of the literature. Obesity 2013, 21, E105–E117.
- Rosmond, R. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology 2005, 30, 1–10.
- Anagnostis, P.; Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009, 94, 2692–2701.
- Stulnig, T.M.; Waldhausl, W. 11beta-Hydroxysteroid dehydrogenase Type 1 in obesity and Type 2 diabetes. Diabetologia 2004, 47, 1–11.
- Duclos, M.; Marquez Pereira, P.; Barat, P.; Gatta, B.; Roger, P. Increased cortisol bioavailability, abdominal obesity, and the metabolic syndrome in obese women. Obes. Res. 2005, 13, 1157–1166.
- Iida, S.; Nakamura, Y.; Fujii, H.; Nishimura, J.; Tsugawa, M.; Gomi, M.; Fukata, J.; Tarui, S.; Moriwaki, K.; Kitani, T. A patient with hypocortisolism and Cushing’s syndrome-like manifestations: Cortisol hyperreactive syndrome. J. Clin. Endocrinol. Metab. 1990, 70, 729–737.
- Friedman, T.C.; Mastorakos, G.; Newman, T.D.; Mullen, N.M.; Horton, E.G.; Costello, R.; Papadopoulos, N.M.; Chrousos, G.P. Carbohydrate and lipid metabolism in endogenous hypercortisolism: Shared features with metabolic syndrome X and NIDDM. Endocr. J. 1996, 43, 645–655.
- Beaupere, C.; Liboz, A.; Feve, B.; Blondeau, B.; Guillemain, G. Molecular Mechanisms of Glucocorticoid-Induced Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 623.
- Scaroni, C.; Zilio, M.; Foti, M.; Boscaro, M. Glucose Metabolism Abnormalities in Cushing Syndrome: From Molecular Basis to Clinical Management. Endocr. Rev. 2017, 38, 189–219.
- Traish, A.M.; Guay, A.T.; Zitzmann, M. 5alpha-Reductase inhibitors alter steroid metabolism and may contribute to insulin resistance, diabetes, metabolic syndrome and vascular disease: A medical hypothesis. Horm. Mol. Biol. Clin. Investig. 2014, 20, 73–80.
- Stewart, P.M.; Krozowski, Z.S. 11 beta-Hydroxysteroid dehydrogenase. Vitam. Horm. 1999, 57, 249–324.
- Tomlinson, J.W.; Stewart, P.M. Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract. Res. Clin. Endocrinol. Metab. 2001, 15, 61–78.
- Napolitano, A.; Voice, M.W.; Edwards, C.R.; Seckl, J.R.; Chapman, K.E. 11Beta-hydroxysteroid dehydrogenase 1 in adipocytes: Expression is differentiation-dependent and hormonally regulated. J. Steroid Biochem. Mol. Biol. 1998, 64, 251–260.
- Janssen, J.; Lamberts, S. Diabetes associated with glucocorticoid excess. In Diabetes Secondary to Endocrine and Pancreatic Disorders; Ghigo, E., Porta, M., Eds.; Karger: Basel, Switzerland, 2014; pp. 22–33.
- Dallman, M.F.; Akana, S.F.; Strack, A.M.; Hanson, E.S.; Sebastian, R.J. The neural network that regulates energy balance is responsive to glucocorticoids and insulin and also regulates HPA axis responsivity at a site proximal to CRF neurons. Ann. N. Y. Acad. Sci. 1995, 771, 730–742.
- Goodman, H.M. Basic Medical Endocrinology, 4th ed.; Elsevier: San Diego, CA, USA, 2009.
- Strack, A.M.; Sebastian, R.J.; Schwartz, M.W.; Dallman, M.F. Glucocorticoids and insulin: Reciprocal signals for energy balance. Am. J. Physiol. 1995, 268, R142–R149.
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12.
- Hansen, K.B.; Vilsboll, T.; Bagger, J.I.; Holst, J.J.; Knop, F.K. Reduced glucose tolerance and insulin resistance induced by steroid treatment, relative physical inactivity, and high-calorie diet impairs the incretin effect in healthy subjects. J. Clin. Endocrinol. Metab. 2010, 95, 3309–3317.