Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Dezhi Mu and Version 2 by Catherine Yang.
Glial cells are the most abundant and widely distributed cells that maintain cerebral homeostasis in the central nervous system. They mainly include microglia, astrocytes, and the oligodendrocyte lineage cells. Moreover, glial cells may induce pathological changes, such as inflammatory responses, demyelination, and disruption of the blood–brain barrier, to regulate the occurrence and development of neurological diseases through various molecular mechanisms.
- RNA m6A modification
- microglia
- astrocyte
- oligodendrocyte
1. Roles of Microglia in Physiological and Pathological States
Microglia are the most important innate immune cells in the CNS. They originate from early yolk sac erythromyeloid progenitors and migrate into the neural tube, where they proliferate and colonize the parenchyma before angiogenesis, as the first glial cells to migrate into the CNS. Macrophages and dendritic cells from the non-cerebral parenchyma (such as the meninges, choroid plexus, and perivascular space) can enter the parenchyma and transform into microglia during embryonic development [1][2][15,16]. Microglial development and maintenance depend on the binding of colony stimulating factor 1 receptor (CSF1R) with colony stimulating factor 1 (CSF1) and IL-34, secreted by neurons and glial cells, and microglia fully mature under the influence of TGF-β secreted by both cells [3][17]. Under normal physiological conditions, microglia, also called “resting microglia,” show the typical branching morphology, with small cell bodies and thin protrusions, and continuously regulate changes in the brain microenvironment by engulfing apoptotic or excess cells and regulating synaptic plasticity and neurogenesis, to maintain the homeostasis of the brain environment. For example, when apoptotic neurons are phosphatidylserine-labeled by growth arrest-specific protein 6 (GAS6), microglia can recognize and phagocytose neurons via the tyro3, axl, and mer (TAM) and milk fat globulin epidermal growth factor 8 (MFGE8) released by astrocytes [4][5][18,19] (Figure 1).
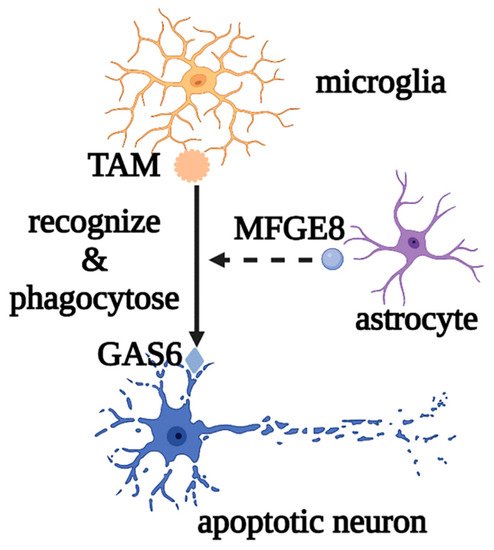
Figure 1. Roles of microglia in phagocytosing apoptotic neurons. Microglia mediate the phagocytosis of apoptotic neurons through the recognition of TAM and GAS6, as well as the auxiliary effect of MFGE8. Abbreviations: GAS6, growth arrest-specific protein 6; TAM, tyro3, axl, and mer; MFGE8, milk fat globulin epidermal growth factor 8.
Microglia also perform synaptic pruning through the direct interaction of the chemokine receptor, CX3CR1, with the transmembrane protein, CX3CL1, on the neuron surface or via the activation of the complement cascade C1q/C3/C3R. Meanwhile, synaptic activity and plasticity can be regulated by reactive oxygen species (ROS), which are secreted by microglia, downregulating the expression of AMPK receptors and brain-derived neurotrophic factor (BDNF)-binding TrkB receptors [6][7][20,21] (Figure 2).
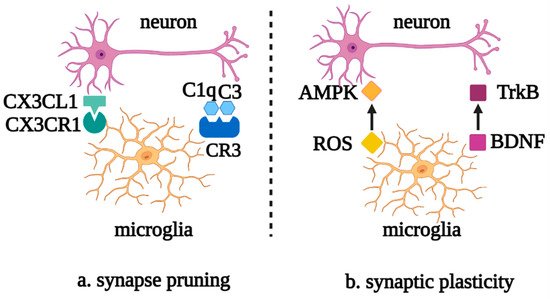
Figure 2. Roles of microglia in neuronal synapse pruning and plasticity. (a) Microglia mediate the neuronal synapse pruning via the complement cascade and the recognition of CX3CR1 and CX3CL1. (b) Microglia mediate synaptic plasticity via the mutual effects of ROS and AMPK, as well as BDNF and TrkB. Abbreviations: CX3CL1, C-X3-C motif chemokine ligand 1; CX3CR1, C-X3-C motif chemokine receptor 1; C1q, complement 1q; C3, complement 3; CR3, complement receptor 3; AMPK, AMP-activated protein kinase; ROS, reactive oxygen species; TrkB, tyrosine kinase receptor B; BDNF, brain-derived neurotrophic factor.
Microglia can also secrete insulin-like growth factor 1 (IGF-1), IL-4, and nitric oxide (NO), promoting the differentiation of neural progenitor cells (NPCs) into oligodendrocytes (OLs) and regulating neurogenesis [8][22].
In addition, as neuroinflammation is one of the common features of CNS pathology, microglia are involved in the regulation of the pathological processes in both acute inflammation in acute injury and chronic inflammation in neurodegeneration [9][10][23,24]. For instance, when induced by Toll-like receptors (TLRs) and other stimuli, microglia are activated with retracted and thickened protrusions and enlarged cell bodies, morphed into short and coarse branching or an ameboid shape, and polarized into M1 and M2 phenotypes according to the pathological damage conditions, time, severity of injury, age, and microenvironment. They participate in antigen presentation, the release of pro-inflammatory/anti-inflammatory cytokines, and the phagocytosis process of necrotic cells. M1 microglia with short and thick branches secrete pro-inflammatory cytokines, such as IL-1β, IL-6, tumor necrosis factor alpha (TNF-α), and neurotoxic substances ROS and NO, which are involved in worsening clinical prognosis in most cases [11][25]. On the other hand, M2 microglia have a typical anti-inflammatory phenotype and ameboid morphology and secrete IL-10, TGF- β, IGF-1, fibroblast growth factor (FGF), CSF1, and neurotrophic factors, to suppress inflammation, engage in regeneration, or mediate phagocytosis [12][26]. In the early stage of inflammation, the excessive adenosine triphosphate (ATP) and glutamate released by damaged neurons activate N-methyl-D-aspartate (NMDA) receptors, which induce excitatory neurotoxicity and lead to neuronal death [13][27]. However, ATP can also cause negative feedback, with the activation of the microglial purinergic receptor P2Y12, inducing the migration of microglia to the injured areas to promote nerve regeneration [14][28]. At the same time, in pathological states, microglia can also engulf pathologically folded proteins, such as tau protein, and myelin sheath fragments, which limits disease progression in the early stage. However, with further development of inflammation, ATP accumulation promotes the activation of the microglial purinergic receptor P2X7 (P2RX7) and NOD-like receptor (NLR) family pyrin domain-containing protein 3 (NLRP3), resulting in the release of excessive pro-inflammatory and neurotoxic factors, which cause phagocytosis dysfunction and brain tissue damage [15][29]. Moreover, microglia can produce prostaglandin D2, which acts on the prostaglandin receptor DP1 of astrocytes, to induce demyelination [2][16]. It can be seen that the morphological changes of microglia in the nervous system are accompanied by corresponding changes in their phenotypes and functions, and each phenotype plays a unique role in the occurrence and development of neurological diseases (Figure 3).
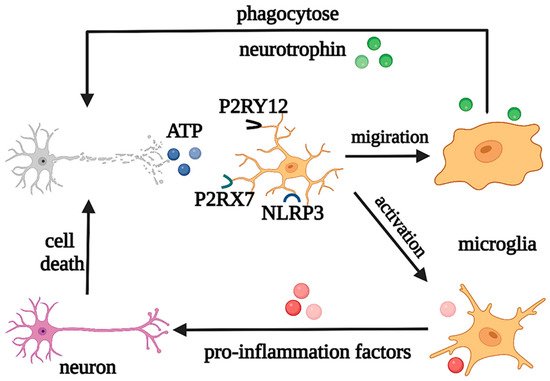
Figure 3. Regulation of microglia under pathological states. Under pathological states, microglia play neuroprotective and neurotoxic roles. Abbreviations: ATP, adenosine triphosphate; P2RY12, purinergic receptor P2Y12; P2RX7, purinergic receptor P2X7; NLRP3, NOD-like receptor (NLR) family pyrin domain-containing protein 3.
2. Roles of Astrocytes in Physiological and Pathological States
Astrocytes are the most widely distributed and numerous glial cells in the mammalian brain. They originate from NPCs and radial glial cells (RGCs) in the subventricular zone, express glial fibrillary acidic protein (GFAP), a specific marker for astrocytes in some regions of the brain, and migrate to the cortical layers, to fill the neuronal space, supporting and dividing neurons. Astrocytes are involved in the regulation of neurotransmitter release and recycling, K+ and Ca2+ balance, synaptogenesis, plasticity maintenance, and BBB homeostasis [16][30]. When stimulated by the K+ and neurotransmitters released by neurons, astrocytes depolarize and trigger the elevation of [Ca2+]i and [K+]i and release of vesicle ATP, regulating the excitability of the axon initial segment of excitatory neurons and the conduction velocity of myelinated axons, thereby controlling neuronal energy metabolism and synaptic numbers [17][18][31,32] (Figure 4).
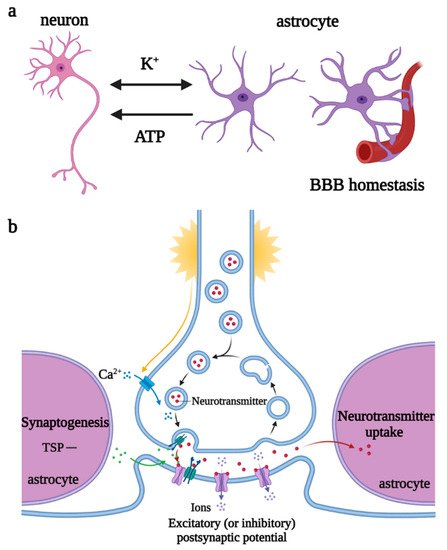
Figure 4. Regulations of astrocytes under physiological states. (a) Astrocytes participate in K+ balance, release ATP to regulate neuronal excitability, and maintain BBB homeostasis. (b) Astrocytes promote synaptogenesis and participate in the regulation of neurotransmitter release and recycling. Abbreviations: ATP, adenosine triphosphate; BBB, blood–brain barrier; TSP, thrombospondin.
Previous studies have shown that, on the one hand, astrocytes undergo reactive hypertrophy and hyperplasia when stimulated by injury, differentiating into A1 neurotoxic and A2 neuroprotective phenotypes. A1 astrocytes promote the production of pro-inflammatory factors and chemokines through the JAK/STAT and NF-κB pathways, resulting in the dysregulation of synaptic release and recycling and morphological changes of synapses and the formation of glial scars under the stimulation of IL-1α, TNF-α, and C1q secreted by microglia [19][20][33,34]. A2 astrocytes can clear ROS and release neurotrophic factors such as BDNF, FGF, nerve growth factor (NGF), and S100 calcium-binding protein A10 (S100A10) to promote neuron survival [21][35]. On the other hand, dysfunctional ion transport in astrocytes leads to neurotoxic cerebral edema, and the morphologic changes in astrocytes cause the endfeet to detach from the vascular surface, destroying the BBB, which allows peripheral blood T cells and monocytes to infiltrate the brain parenchyma and interact with microglia, to exacerbate the inflammatory response [22][23][36,37]. At the same time, astrocyte orosomucoid-2 binds to the C-C chemokine receptor type 5 (CCR5) of microglia to block the CXCL4-CCR5 binding domain, inhibiting microglial activation and migration [24][38]. Astrocytes regulate microglial phagocytic activity via plasminogen activator inhibitor type 1 (PAI-1) release, the low-density lipoprotein receptor-related protein 1 (LRP-1)/JAK/STAT1 axis, and TLR2/6 [25][39] (Figure 5).
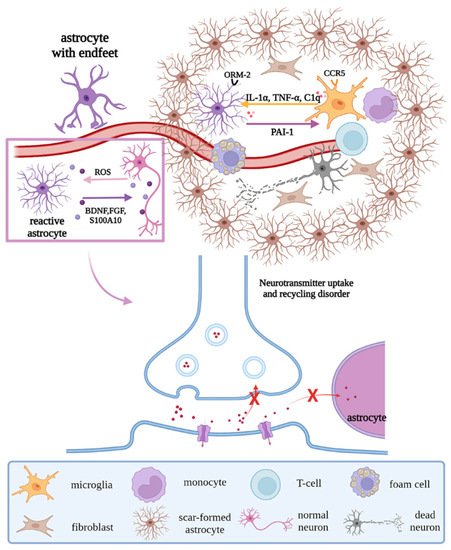
Figure 5. Regulations of astrocytes under pathological states. Under pathological states, reactive astrocytes remove ROS and release neurotrophic factors, to promote the survival of neurons; in addition, their endfeet strip off the vascular surface, resulting in the destruction of the BBB and dysregulation of synaptic release and recycling, with morphological changes. Moreover, under the interaction with microglia and stimulation of factors secreted by microglia, scar-formed astrocytes, peripheral immune cells, and fibroblasts form glial scar. Abbreviations: ROS, reactive oxygen species; BDNF, brain-derived neurotrophic factor; FGF, fibroblast growth factor; S100A10, S100 calcium binding protein A10; ORM-2, orosomucoid-2; CCR5, C-C chemokine receptor type 5; C1q, complement 1q; PAI-1, plasminogen activator inhibitor type 1; “×”, the infeasibility of the approach.
3. Roles of Oligodendrocytes in Physiological and Pathological States
Oligodendrocyte lineage cells are the myelin-forming cells in the CNS, which mainly include the oligodendrocyte progenitor cells (OPCs), pre-OLs, and OLs. Arising from NPCs and subsisting during the process of development, OPCs, also known as NG2 glial cells (whose markers also include oligodendrocyte transcription factor (Olig2) and SRY-box transcription factor 10 (SOX10)), have a bipolar morphology and maintain a highly dynamic balance between proliferation and differentiation. OPCs migrate to myelination sites via the WNT pathway, to crawl or jump along blood vessels, and continuously produce bipolar pre-OLs throughout their life cycle [26][27][28][40,41,42]. When in contact with axons, the bipolar morphology of pre-OLs changes, and pre-OLs differentiate into mature OLs (whose markers include myelin associated glycoprotein (MAG), myelin basic protein (MBP), proteolipid protein (PLP), and 2′, 3′-cyclic nucleotide 3′phosphodiesterase (CNPase)). The cytoplasm of OLs wraps axons and forms a myelin sheath for rapid action potential transmission, owing to the high resistance and low capacitance of the sheath and the existence of the nodes of Ranvier. OLs can also provide nutritional and metabolic support for axons by transporting lactate and ATP [29][43].
OLs are noted as crucial cells for myelination, and mature OL deficiency may cause axonal loss and demyelination, leading to brain injury, autoimmune diseases, demyelinating or chronic degenerative diseases, and other myelination disorders. Under normal physiological conditions, a deficiency of OLs induces the differentiation of OPCs into new OLs and migration to the lesion sites, under the secretion of factors by microglia and astrocytes, such as BDNF and FGF, to restart the myelination process and cover the injured sites or axons that have not previously been myelinated [30][44]. During myelin regeneration, the transcription factors required for OPC differentiation, such as NK2 homeobox 2 (NKX2.2), myelin regulatory factor (MYRF), zinc finger protein 488 (ZFP488), Olig1, and Olig2, are highly expressed, to initiate myelinogenesis. However, pathological studies of most demyelinating diseases have shown that OPCs are the main damaged cells, including the obstruction of differentiation of OPCs to OLs (e.g., white matter injury diseases), inhibition of OPC migration to the injured sites (e.g., multiple sclerosis), and ultimately increased OPC apoptosis (e.g., senescence). Therefore, the functions of OL lineage cells change through a variety of molecular mechanisms, further regulating the occurrence and development of various neurological diseases [31][32][45,46] (Figure 6).
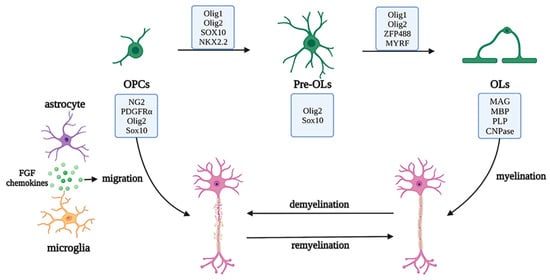
Figure 6. Development of the oligodendrocyte lineage and migration and regeneration under pathological conditions. The oligodendrocyte lineage is composed of the OPCs, pre-OLs, and OLs with unique markers and OLs cytoplasm, which wraps axons to form myelin sheaths. Under pathological conditions, the myelin is damaged, the neurons are demyelinated, and myelin regeneration is initiated by OPC activation and migration to injured sites, with the assistance of astrocytes and microglia and a series of transcription factors. Abbreviations: PDGFRα, platelet derived growth factor receptor alpha; SOX10, SRY-box transcription factor 10; NKX2.2, NK2 homeobox 2; Olig1/2, Oligodendrocyte transcription factor 1/2; ZFP488, zinc finger protein 488; MYRF, myelin regulatory factor; MAG, myelin associated glycoprotein; MBP, myelin basic protein; PLP, proteolipid protein; CNPase, 2′,3′-cyclic nucleotide 3′phosphodiesterase; OPC, oligodendrocyte progenitor cells; OL, oligodendrocytes.