Nearly half a century has passed since the discovery of cytoplasmic inheritance of human chloramphenicol resistance. The inheritance was then revealed to take place maternally by mitochondrial DNA (mtDNA) and mutations in mtDNA were identified as a cause of severe inheritable metabolic diseases with neurological manifestation. A growing number of preclinical studies have revealed that animal behaviors are influenced by the impairment of mitochondrial functions. Indeed, as high as 54% of patients with one of the most common primary mitochondrial diseases, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome, present psychiatric symptoms. Mitochondrial functions are observed to be compromised and to become less resilient under continuous stress. Furthermore, stress, inflammation, mitochondrial impairment have been linked to the activation of the tryptophan–kynurenine metabolic system, which observably contributes to the development of pathological conditions including neurological and psychiatric disorders.
- mitochondria
- stress resilience
- plasticity
- stress
- kynurenine
- neurodegenerative
- psychiatry
- Alzheimer's disease
- depression
- anxiety
1. Introduction
2. Mitochondria in the Central Nervous System
The brain accounts for only 2% of the body weight; however, it consumes as much as 20% of body’s total oxygen supply. An estimated number of one to two million mitochondria is present per single neuron in the human substantia nigra [28]. Mitochondria take responsibility for the production of cellular energy and the proper conduction of neural circuits in the nervous system [29][30][31][32][33]. Mitochondria are multifunctional organelles maintaining calcium homeostasis and signaling to other organelles in the cell as well as with other mitochondria at distance [34][35]. Furthermore, mitochondria are highly plastic in morphology, functions, and cell cycle, depending on the tissue type and the need of cells [36]. Mitochondria can be even transferred from cell to cell [37].2.1. Mitochondrial Bioenergetics
Glucose, other sugars, and some amino acids are broken down in the cytosol to three-carbon molecule pyruvate which transfers into the mitochondria. Pyruvate is degraded to two carbon molecule acetyl coenzyme A (acetyl-CoA) which enters the second stage of cellular respiration, the TCA cycle that takes place in the matrix of mitochondria. Initially, Szent-Györgyi reported a cyclic chemical reaction between a four-carbon molecule (oxaloacetate) and two four-carbon molecules (fumarate and L-(-)-malate (the Szent-Györgyi cycle)). Later, Krebs revealed larger cyclic biochemical reactions in which a two-carbon molecule “triose” bonds with oxaloacetate to form a six-carbon molecule citrate, which is then oxidized to a five-carbon molecule (alpha-ketoglutarate) and four-carbon molecules (succinyl-CoA, succinate, fumarate, and malate), thus forming the TCA cycle (the Krebs cycle) [38]. “Triose” was eventually identified as a product of pyruvate and coenzyme A, acetyl-CoA [39][40]. The TCA cycle employs eight different enzymes, reproducing one molecule of oxaloacetate, two molecules of carbon dioxide, water, three molecules of NADH, and one molecule of flavin adenine dinucleotide (FADH2) and guanosine triphosphate (GTP). GTP is readily converted to ATP. In the TCA cycle, most of the high-energy storage molecule ATP is consumed by NAD+ and FAD to form NADH and FADH2 [29] (Figure 1a). The NAD+ excess has been reported to improve mitochondrial function and thus prolong the life span of mice [30].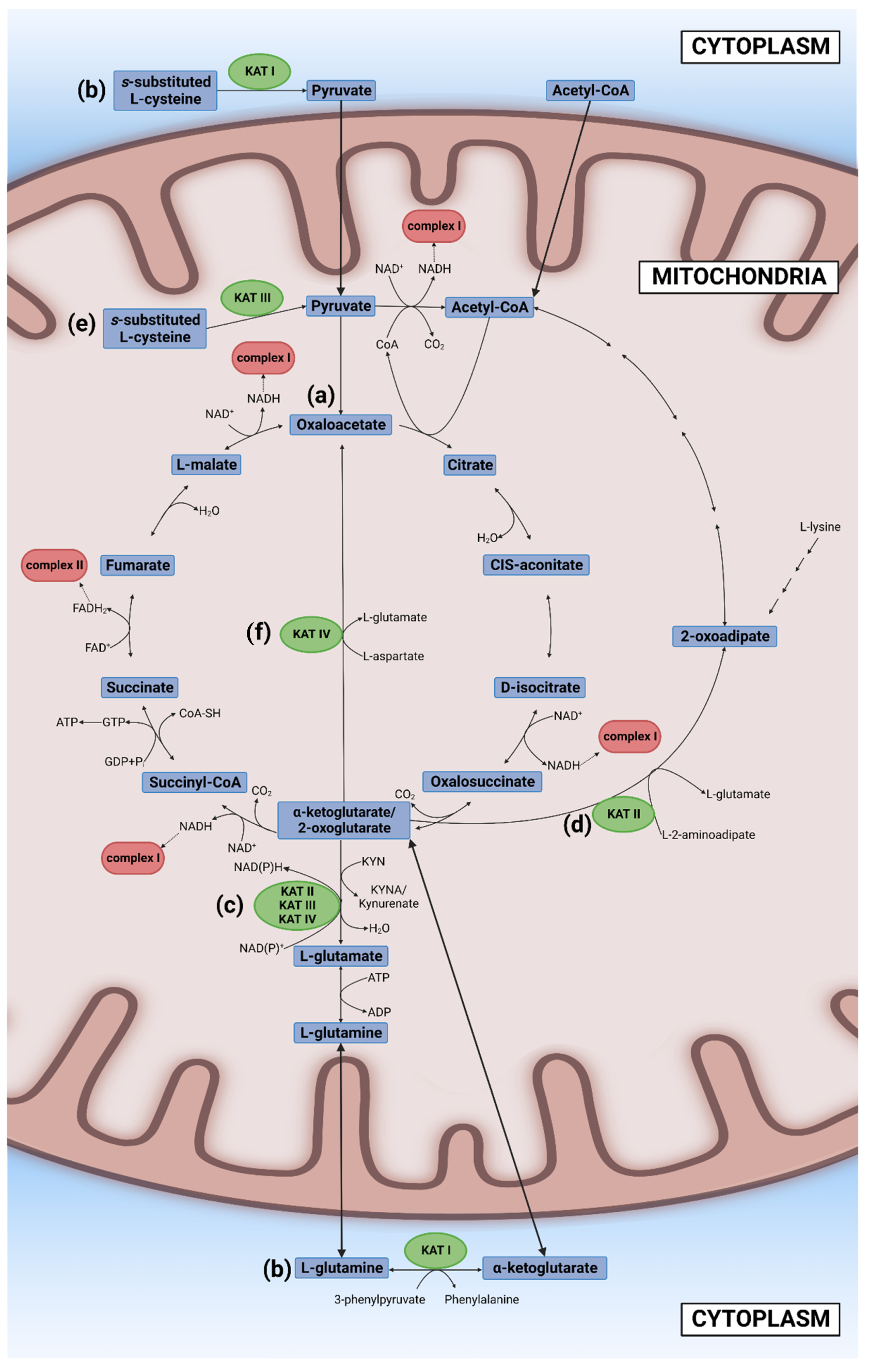
2.2. Other Mitochondrial Functions
Mitochondria play an important role in cellular calcium homeostasis. The concentration of calcium ions in the intermembrane space is the same as that in the cytosol due to the high permeability of the outer mitochondria membrane. A higher concentration of mitochondrial calcium ions enhances ATP production; however, severe calcium overloads are associated with pathological conditions [34][35][41]. Mitochondria constantly communicate with other cellular organelles such the nucleus, the ER, lysozymes, and peroxisomes. The coordinated interaction of mitochondrial and nuclear factors is required for mitochondrial gene expression offered by mitochondrial ribonuclease P, ribosomal RNAs, transfer RNAs, introns, and a protein [42]. The nucleus sends signals to the mitochondria via anterograde regulation to modulate mitochondrial biogenesis upon stressful events. On the other hand, mitochondria constantly transmit information on mitochondrial status and cellular stress to the nucleus by retrograde signaling [43]. Mitochondria and the ER are at a close contact through the mitochondria-associated membrane to exchange information on energy production, calcium homeostasis, lipid transport, and apoptosis [44]. Lysosomes interact with mitochondria to transport amino acids, lipids, and calcium ions [45]. Mitochondria and peroxisomes function in concert in fatty acid metabolism. Mitochondria degrade long-chain fatty acids to supply acetyl-CoA and produce ATP, while peroxisome performs beta-oxidation to generate hydrogen peroxide and anabolic metabolic metabolism such as plasmalogen and bile acid synthesis [46]. Mitochondria undergo division during mitosis, dividing equally between the cell soma to daughter cells in interaction with the ER and cytoskeleton [47]. The morphology, functions, and dynamics of mitochondria change upon tissue differentiation [48]. Mitochondria constantly divide and fuse, controlling their morphology and functions. The fusion takes place by initially merging the outer membrane and subsequently the inner membrane of two mitochondria. The continuous events of fusion and division generate mitochondrial networks [49]. Mitophagy refers to mitochondrial autophagy in which double-membraned vesicle autophagosomes deliver mitochondria to lysosomes for destruction. Mitophagy is induced by prolonged fission, promoting the repair process, but may lead to mitochondrial degradation. MicroRNAs play an important role in regulation of protein expression responsible for autophagy [50]. Mitochondria also induce an immune response via the activation of the mitochondrial antiviral signaling protein which leads to the secretion of cytokines via the virally infected cells [51]. Furthermore, mitochondria induce mitochondrial apoptosis through mitochondrial outer membrane permeabilization which leads to the disruption of mitochondrial outer membrane and the release of intermembrane space proteins such as cytochrome c [52]. Therefore, mitochondria impairment may lead to multifarious consequences from ion homeostasis to entire organismal levels.References
- Fiske, C.H.; Subbarow, Y. Phosphorus Compounds of Muscle and Liver. Science 1929, 70, 381–382.
- Lohmann, K. Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften 1929, 17, 624–625.
- Maruyama, K. The Discovery of Adenosine Triphosphate and the Establishment of its Structure. J. Hist. Biol. 1991, 24, 145–154.
- Krebs, H.A.; Johnson, W.A. Metabolism of ketonic acids in animal tissues. Biochem. J. 1937, 31, 645–660.
- Friedkin, M.; Lehninger, A.L. Esterification of inorganic phosphate coupled to electron transport between dihydrodiphosphopyridine nucleotide and oxygen. J. Biol. Chem. 1949, 178, 611–644.
- Belitser, V.A.; Tsibakova, E.T. About phosphorilation mechanism coupled with respiration. Biokhimiya 1939, 4, 516–534.
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228.
- Knoop, F. Der Abbau aromatischer Fettsäuren im Tierkörper. Beitr. Chem. Physiol. Pathol. 1904, 6, 150–162.
- Klingman, D.; Handler, P. Partial purification and properties of renal glutaminase. J. Biol. Chem. 1958, 232, 369–380.
- Ernster, L.; Ikkos, D.; Luft, R. Enzymatic activities of human skeletal muscle mitochondria: A tool in clinical metabolic research. Nature 1959, 184, 1851–1854.
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719.
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430.
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995, 11, 144–149.
- Carafoli, E. The interplay of mitochondria with calcium: An historical appraisal. Cell Calcium 2012, 52, 1–8.
- Diogo, C.V.; Yambire, K.F.; Fernández Mosquera, L.; Branco, F.T.; Raimundo, N. Mitochondrial adventures at the organelle society. Biochem. Biophys. Res. Commun. 2018, 500, 87–93.
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431.
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734.
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564.
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778.
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784.
- Koene, S.; Wortmann, S.B.; de Vries, M.C.; Jonckheere, A.I.; Morava, E.; de Groot, I.J.; Smeitink, J.A. Developing outcome measures for pediatric mitochondrial disorders: Which complaints and limitations are most burdensome to patients and their parents? Mitochondrion 2013, 13, 15–24.
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137.
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress and Mitochondrial Damage in Neurodegenerative Diseases: From Molecular Mechanisms to Targeted Therapies. Oxid. Med. Cell Longev. 2020, 2020, 1270256.
- Rigotto, G.; Basso, E. Mitochondrial Dysfunctions: A Thread Sewing Together Alzheimer’s Disease, Diabetes, and Obesity. Oxid. Med. Cell Longev. 2019, 2019, 7210892.
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344.
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824.
- Daniels, T.E.; Olsen, E.M.; Tyrka, A.R. Stress and Psychiatric Disorders: The Role of Mitochondria. Annu. Rev. Clin. Psychol. 2020, 16, 165–186.
- Raichle, M.E.; Gusnard, D.A. Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. USA 2002, 99, 10237–10239.
- Kay, J.; Weitzman, P.D. Krebs’ Citric Acid Cycle: Half a Century and Still Turning; Biochemical Society: London, UK, 1987; p. 25.
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443.
- Stryer, L. Fatty acid metabolism. In Biochemistry, 4th ed.; W.H. Freeman and Company: New York, NY, USA, 1995; pp. 603–628.
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526105/ (accessed on 6 July 2022).
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021.
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819.
- Xu, Z.; Zhang, D.; He, X.; Huang, Y.; Shao, H. Transport of Calcium Ions into Mitochondria. Curr. Genom. 2016, 17, 215–219.
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 8952520.
- Liu, D.; Gao, Y.; Liu, J.; Huang, Y.; Yin, J.; Feng, Y.; Shi, L.; Meloni, B.P.; Zhang, C.; Zheng, M.; et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal. Transduct. Target Ther. 2021, 6, 65.
- Krebs, H.A. The citric acid cycle and the Szent-Györgyi cycle in pigeon breast muscle. Biochem. J. 1940, 34, 775–779.
- Lipmann, F.; Kaplan, N.O. A common factor in the enzymatic acetylation of sulfanilamide and of choline. J. Biol. Chem. 1946, 162, 743–744.
- Lipmann, F. Development of the Acetylation Problem: A personal Account. Available online: https://www.nobelprize.org/prizes/medicine/1953/lipmann/lecture/ (accessed on 6 July 2022).
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167.
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27.
- Soledad, R.B.; Charles, S.; Samarjit, D. The secret messages between mitochondria and nucleus in muscle cell biology. Arch. Biochem. Biophys. 2019, 666, 52–62.
- Yeo, A.J.; Chong, K.L.; Gatei, M.; Zou, D.; Stewart, R.; Withey, S.; Wolvetang, E.; Parton, R.G.; Brown, A.D.; Kastan, M.B.; et al. Impaired endoplasmic reticulum-mitochondrial signaling in ataxia-telangiectasia. iScience 2020, 24, 101972.
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and Lysosomes: Discovering Bonds. Front. Cell Dev. Biol. 2017, 7, 106.
- Demarquoy, J.; Le Borgne, F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 2015, 6, 301–319.
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646.
- Noguchi, M.; Kasahara, A. Mitochondrial dynamics coordinate cell differentiation. Biochem. Biophys. Res. Commun. 2018, 27, 59–64.
- Hales, K.G. Mitochondrial Fusion and Division. Nat. Educ. 2010, 3, 12.
- Thomas, R.L.; Gustafsson, A.B. Mitochondrial autophagy—An essential quality control mechanism for myocardial homeostasis. Circ. J. 2013, 77, 2449–2454.
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial Interactome: A Focus on Antiviral Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 8.
- Vringer, E.; Tait, S.W.G. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Biol. 2019, 7, 100.