Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Natalia A. Shnayder.
Among neurological adverse drug reactions (ADRs) in patients with schizophrenia treated with antipsychotics (APs), drug-induced parkinsonism (DIP) is the most common motility disorder caused by drugs affecting dopamine receptors. One of the causes of DIP is the disruption of neurotransmitter interactions that regulate the signaling pathways of the dopaminergic, cholinergic, GABAergic, adenosinergic, endocannabinoid, and other neurotransmitter systems. Presently, the development mechanisms remain poorly understood despite the presence of the considered theories of antipsychotic-induced parkinsonism (AIP) pathophysiology.
- antipsychotic-induced parkinsonism
- drug-induced parkinsonism
- antipsychotics
- dopaminergic system
- dopaminergic receptor
- adverse drug reaction
1. Dopamine D2 Type Receptors Blockade
Dopamine receptors in the brain are represented by two families: the D1 (D1 family receptors and D5 receptors), and the D2 (D2, D3 and D4 receptors). All currently available APs are able to antagonize dopamine D2 receptors, and the APs’ therapeutic effects in psychosis are related to their action on the limbic system reducing dopamine transmission. Due to antagonism of dopamine D2 receptors in the striatum, neurons of gamma aminobutyric acid (GABA), encephalin and the subthalamic nucleus are disinhibited at the beginning of the indirect pathway without changing the direct pathway. Due to this, there is an increase in GABA inhibition in the thalamocortical projection by facilitating inhibition in the globus pallidus / reticular substantia nigra. This pathway is similar to the model of basal ganglia motor loop impairment in Parkinson disease (PD) [4][1].
It is assumed that the mechanism of action of APs is associated with the level of occupancy of the dopamine D2 receptors. This is confirmed by several reports that therapeutic doses of typical APs block D2 receptors in 70–89% of cases in young adults, while atypical APs block in 38–63% of cases [10][2].
2. Supersaturation (“Occupancies”) of Striatal Dopamine D2/3 Receptors
The exact mechanism of AIP development is still unknown; nevertheless, the main theory is the dopamine receptors blockade. In animal models, about 70% of the occupancy of dopamine D2/3 receptors was recorded during AP therapy, which leads to the development of AIP [11][3]. The threshold levels of occupancy of dopamine D2/3 receptors in the striatum associated with the development of AIP in young adults, which is about 80%, have been demonstrated in studies using neuroimaging technologies (using positron emission tomography (PET) D2/3 receptor imaging) [12,13][4][5]. Dissimilarities in the severity of AIP development are associated with the occupancy density of dopamine D2/3 receptors, AP concentration, the rate of dissociation from the D2 receptor, the selectivity of dopamine receptors in the limbic system and striatum, and the activity of other receptors (for example, serotonergic, muscarinic) [14][6]. Therefore, typical APs are more associated with an increased risk of developing AIP than atypical APs [14,15,16][6][7][8]. However, such a high occupancy of dopamine receptors should not be considered unequivocally, because the occupancy of receptors is not equal to antagonism. For example, aripiprazole, which, in addition to dopamine receptors, interacts with serotonin (5-HT) types 1A and 2A receptors and rarely causes AIP, even with a dopamine receptor occupancy rate > 95% due to a weak antagonistic effect on dopamine receptors [17,18][9][10].
3. Influence of the Basal Ganglia of the Thalamocortical Motor Loop
The pathophysiology of AIP is associated with drug-induced changes in the motor chain of the basal ganglia secondary to blockade of dopaminergic receptors [4][1]. The central dopaminergic system is presented of the four pathways: mesolimbic, mesocortical, tuberoinfundibular, and nigrostriatal (Figure 21).
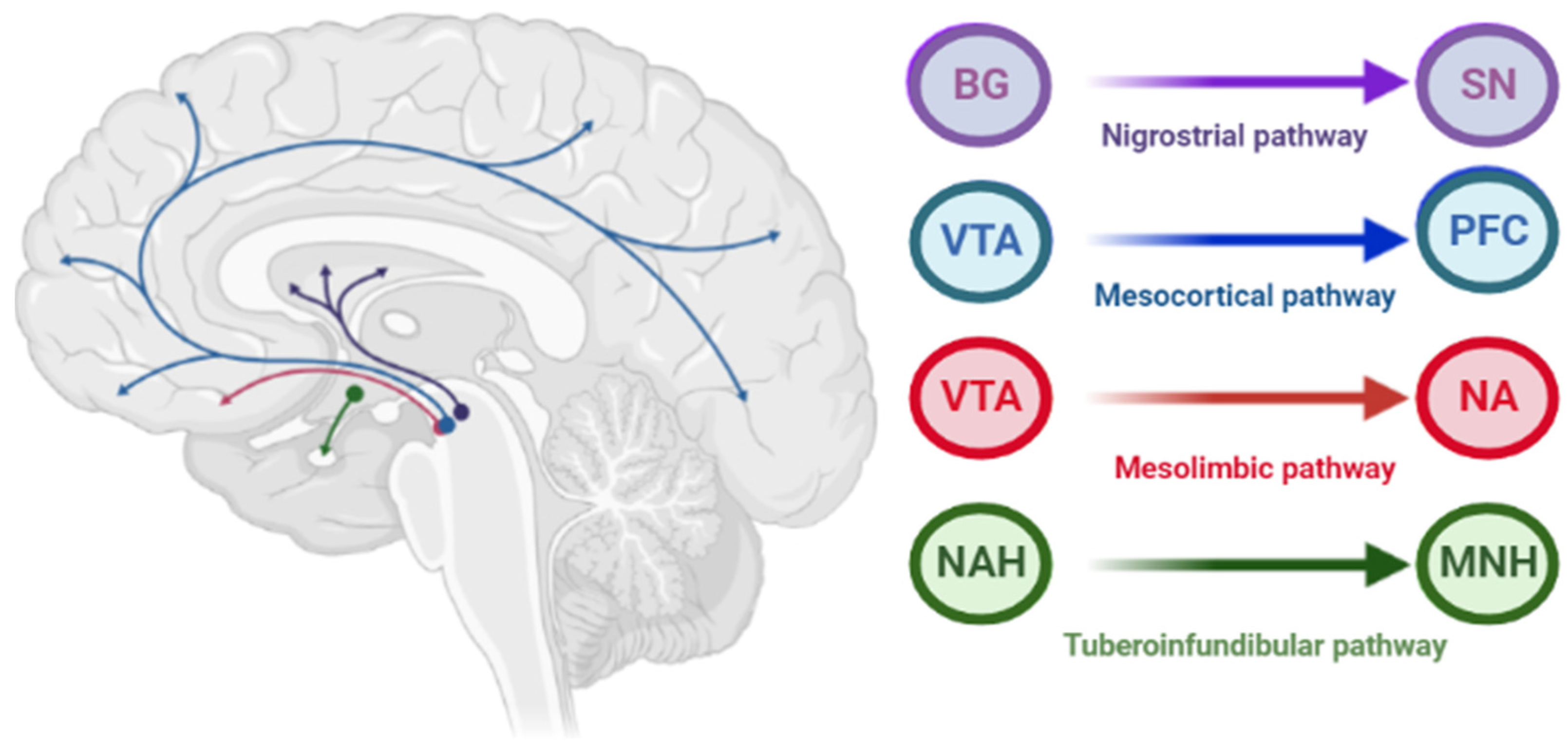
Figure 21. Pathways of dopaminergic neurotransmission. Note: VTA—ventral tegmental area; PFC—prefrontal cortex; NA—nucleus accumbens: SN—substance nigra; BG—basal ganglia (striatum); NAH—nucleus arcuatus (hypothalamus); MNH—middle nucleus (hypothalamus).
When dopamine D2 receptors blockade in the striatum, striatal neurons containing GABA and encephalin are disinhibited, affecting the indirect pathway and ultimately leading to a relative decrease in the activity of thalamocortical circuits [4,19][1][11]. This effect may be mitigated by APs anticholinergic activity [20[12][13],21], as evidenced by the observation that clozapine has a low risk of developing AIP and also has a high relative affinity for muscarinic cholinergic receptors [20][12]. The reason for the decrease in sufficient concentrations of dopamine in the striatum may also be a decrease in the release of dopamine into the synaptic cleft [21][13]. Drugs that do not directly affect dopamine levels (valproic acid, calcium channel blockers) can induce DIP through other mechanisms, including modulation of GABA activity or mitochondrial dysfunction (Figure 32) [4,21,22,23,24][1][13][14][15][16].
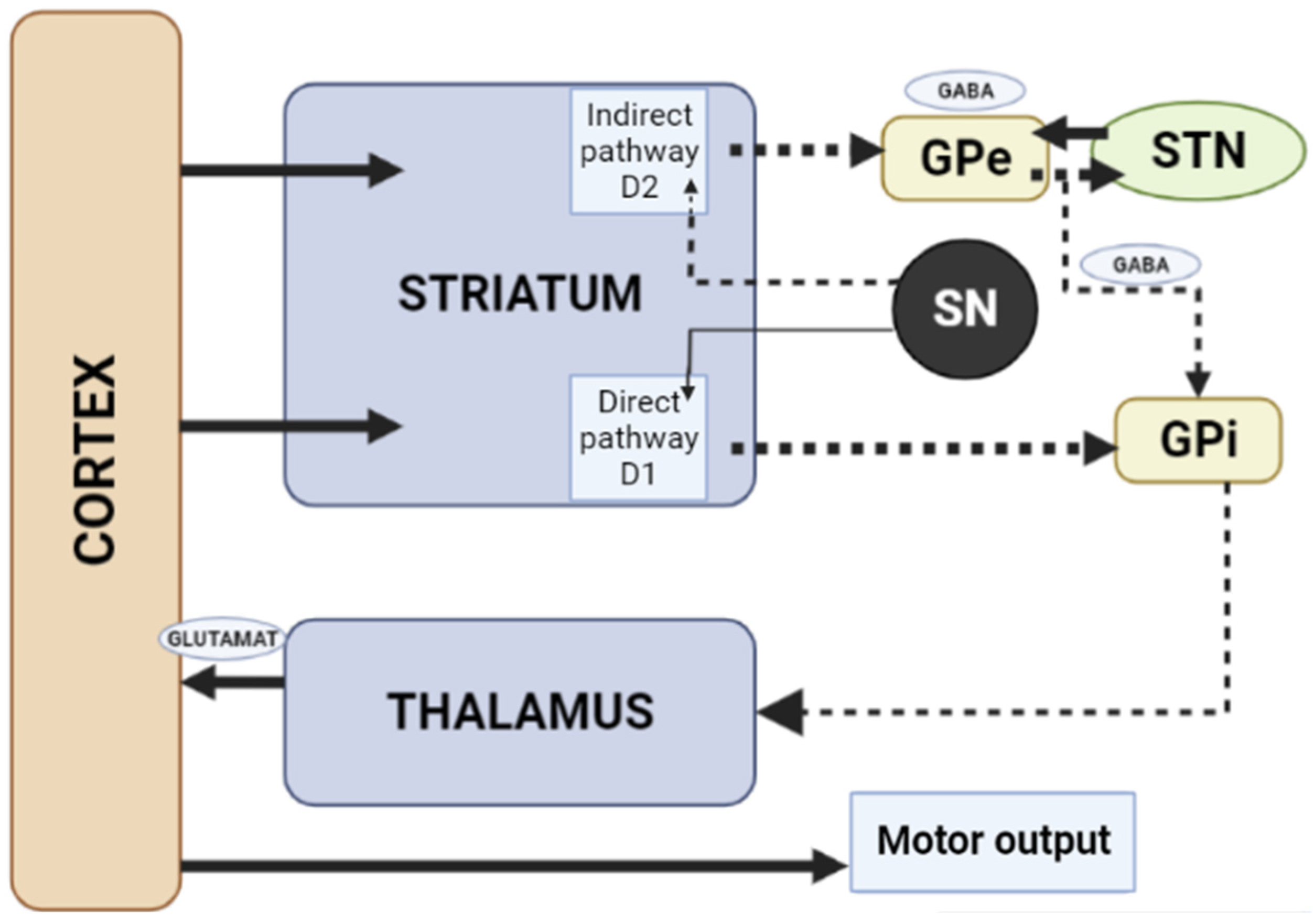
Figure 32. Schematic diagram of the excitatory and inhibitory ganglia involved in the development of AIP (Adapted from [25][17], Copyright year 2020, BMJ Neurol. Open). Note: Substantia nigra dopaminergic projections exert an exciting effect on stria-pallidal fibers of the direct pathway through dopamine D1 receptors, which leads to disinhibition of the thalamic nuclei and increased thalamocortical excitation, facilitating movements initiated by the cortex. The obstruction of voluntary movement occurs due to thalamic inhibition, due to the inhibition of stria-pallidal fibers in an indirect pathway through dopamine D2 receptors. The direct pathway is due to the activation of glutamate neurons in the sensorimotor cortex, and the indirect pathway is due to the activation of GABA-ergic neurons. The dotted line shows the inhibitory action due to the action of GABA. The straight line shows the excitatory effect due to the action of glutamate.
4. Fast-off-D Theory
In studies devoted to brain occupancy, radioactive clozapine has been proven to show rapid and transient occupancy of the dopamine D2 type receptors, dissociating in less than 60 s after administration, while radioactive haloperidol and chlorpromazine show long-term occupancy with slow dissociation in less than 30 min. Therefore, atypical APs are clinically more effective, having temporary occupancy of D2 type dopamine receptors and rapid dissociation to normal dopamine neurotransmission [26,27][18][19].
5. Role of Adenosine Receptors
Purine and adenosine interact with major neurotransmitter systems (glutamatergic cholinergic, GABA-ergic and dopaminergic) to modulate neuronal function in the central and peripheral nervous systems [28][20].
Transmission of adenosine occurs through purinergic receptors coupled to the G-protein class P1, which is subdivided into four receptor subtypes: A1, A2A, A2B, and A3 [29][21]. A2A adenosine receptors are highly expressed by GABAergic neurons in the striatum, globus pallidus, and olfactory bulb [30][22], and are co-localized with dopamine D2 type receptors in the basal ganglia on enkephalin-expressing output neurons of the indirect pathway leading to the globus pallidus and substantia nigra [31[23][24],32], which are in dopaminergic nigrostriatal and mesolimbic neuronal pathways [33][25]. The A2A and D2 receptors are antagonists and regulate GABA neurons [34,35][26][27].
Most evidence suggests that due to intramembrane interaction, activation of the adenosine A2A receptor indirectly blocks the activation of dopamine D2 receptors, and stimulation of D2 receptors blocks the activation of adenylate cyclase caused by the A2A receptor [34][26]. Upon stimulation of A2A receptors, GABA is released, while upon stimulation of D2 receptors, it is suppressed in the globus pallidus [35,36][27][28]. Here is evidence that these receptors, on the contrary, can act as synergists, under certain circumstances (presence of certain isoforms of adenylate cyclase, interruption of previous long-term exposure to D2 receptor agonists), activation of the D2 receptors enhances the effects of A2A receptors [34][26]. A study by Parsons et al. [37][29] in rats showed that chronic administration of haloperidol activates striatal A2A receptors. The effect of haloperidol was selective for A2A receptors over other adenosine receptor subtypes. Notably, atypical APs did not affect A2A receptors’ density in this study [37][29]. A2A adenosine receptor antagonists suppress motor disorders such as catalepsy and locomotion induced by dopamine antagonists [38,39][30][31]. A2A receptor antagonists are effective in relieving muscle rigidity and tremor in AIP (Figure 43) [15,40,41][7][32][33].
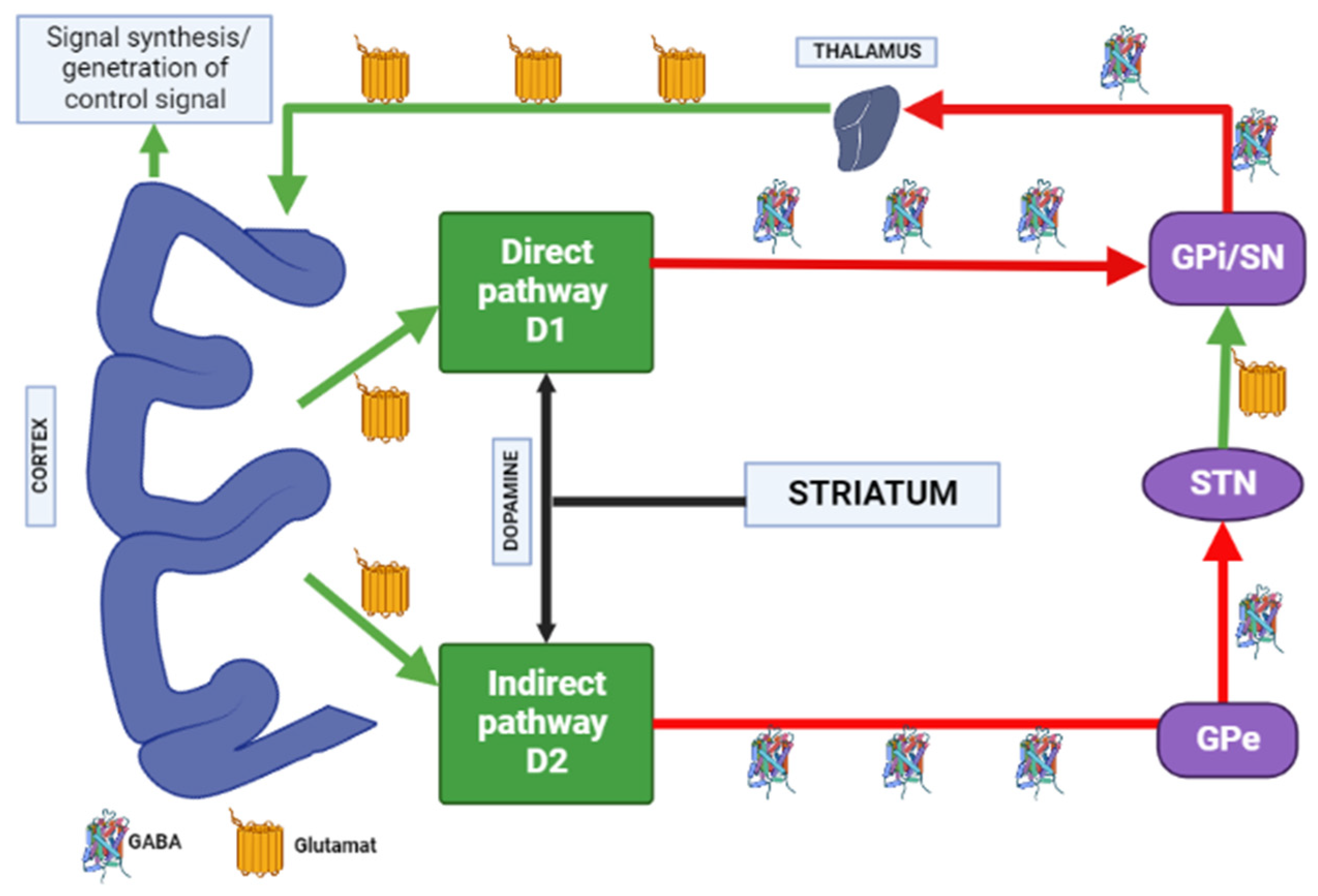
Figure 43. Distribution of A1 and A2A adenosine receptors in the human brain. Note: GPe—globus pallidus external; GPi—globus pallidus internal; STN—subthalamic nucleus; SN—substantia nigra; GABA—gamma aminobutyric acid.
6. Blockade of the Serotonergic System
The serotonergic system plays a crucial role in various physiological actions regulation, including psychoemotional, cognitive, sensorimotor, and autonomic functions [42,43,44][34][35][36]. Serotonergic neurotransmission is presented by several 5-HT receptors, which are classified into 7 families (5-HT1 to 5-HT7) and 14 subtypes (5-HT1A, 1B, 1D, 1E, 1F, 5-HT2A, 2B, 2C, 5-HT4, 5-HT3, 5-HT5A, 5B, 5-HT6 and 5-HT7) [45,46][37][38]. The 5-HT1E, 5-HT1F, and 5-HT5 receptors are associated with the Gi/o protein and inhibit adenylate cyclase activity, cyclic adenosine monophosphate (cAMP) formation, and protein kinase A (PKA) activity. The 5-HT2A, 5-HT2B, and 5-HT2C receptors are coupled to the Gq protein and increase phosphatidylinositol (PI) turnover by activating phospholipase C, thereby stimulating the protein kinase C− and Ca2+/calmodulin cascade. The 5-HT4, 5-HT6, and 5-HT7 receptors are Gs-coupled and activate adenylate cyclase and PKA [44,45,46][36][37][38].
5-HT1A receptors are localized in the raphe nucleus, hippocampus, amygdala, and lateral septum. Expression of 5-HT1A receptors occurs in the cerebral cortex, basal ganglia (striatum), and diencephalon (thalamus and hypothalamus) in low to moderate density [42,47,48][34][39][40]. Inhibition of adenylate cyclase activity by 5-HT1A receptors leads to inhibition of the cAMP-PKA cascade. In addition, 5-HT1A receptors activate G-protein-gated inwardly rectifying potassium channels (GIRK), which hyperpolarize target neurons and suppress their activity [41,43,44,45][33][35][36][37]. Several studies on animal models have shown that 5-HT1A receptors are activated with the introduction of AP for dehydration, has a protective effect on the development of extrapyramidal syndrome (EPS) [49,50,51,52,53,54,55][41][42][43][44][45][46][47]. The 5-HT2A and 5-HT2C receptors are highly expressed in the cerebral cortex, olfactory tubercle and limbic system (nucleus accumbens, hippocampus), basal ganglia (striatum and substantia nigra). 5-HT2A/2C antagonists attenuate AP-induced EPS in PD patients [56,57,58][48][49][50] by increasing the level of released acetylcholine, dopamine metabolism, and Fos protein expression in the striatum [57][49]. 5-HT3 receptors constitute a heteropentamer consisting of subunits from 5-HT3A to 5-HT3E and function as a cation (Na+, K+, and Ca2+) permeable channels [45,59][37][51]. Therefore, when 5-HT3 receptors are activated, postsynaptic membranes are depolarized, thereby exciting target neurons. Serotonin 5-HT3 receptors are located in the nerve endings of various neurons and induce the release of neurotransmitters (acetylcholine, glutamate, GABA and dopamine). Clinical studies have also shown that 5-HT3 receptor antagonists significantly reduce the frequency and severity of AP-induced EPS (e.g., AIP) in patients with chronic schizophrenia [60,61][52][53]. However, a recent study showed that serotonin 5-HT3 receptors do not affect the activity of cholinergic interneurons in the striatum [62][54]. Thus, the functional mechanisms of 5-HT3 receptors in EPS are still unclear. The expression of serotonin 5-HT6 receptors mainly occurs in the brain, in particular, in the basal ganglia (striatum and nucleus accumbens), limbic system (olfactory tubercles and hippocampus), and cerebral cortex [45][37]. According to the results of clinical studies, 5-HT6 receptor antagonists also have a protective property against the development of EPS [63,64][55][56]. The decrease in the incidence and severity of EPS in the presence of 5-HT6 antagonists was further confirmed by an electrophysiological study [62][54], reflecting a decrease in the activation of striatal acetylcholine neurons, thereby reducing the likelihood of developing EPS (Figure 54) [65][57].
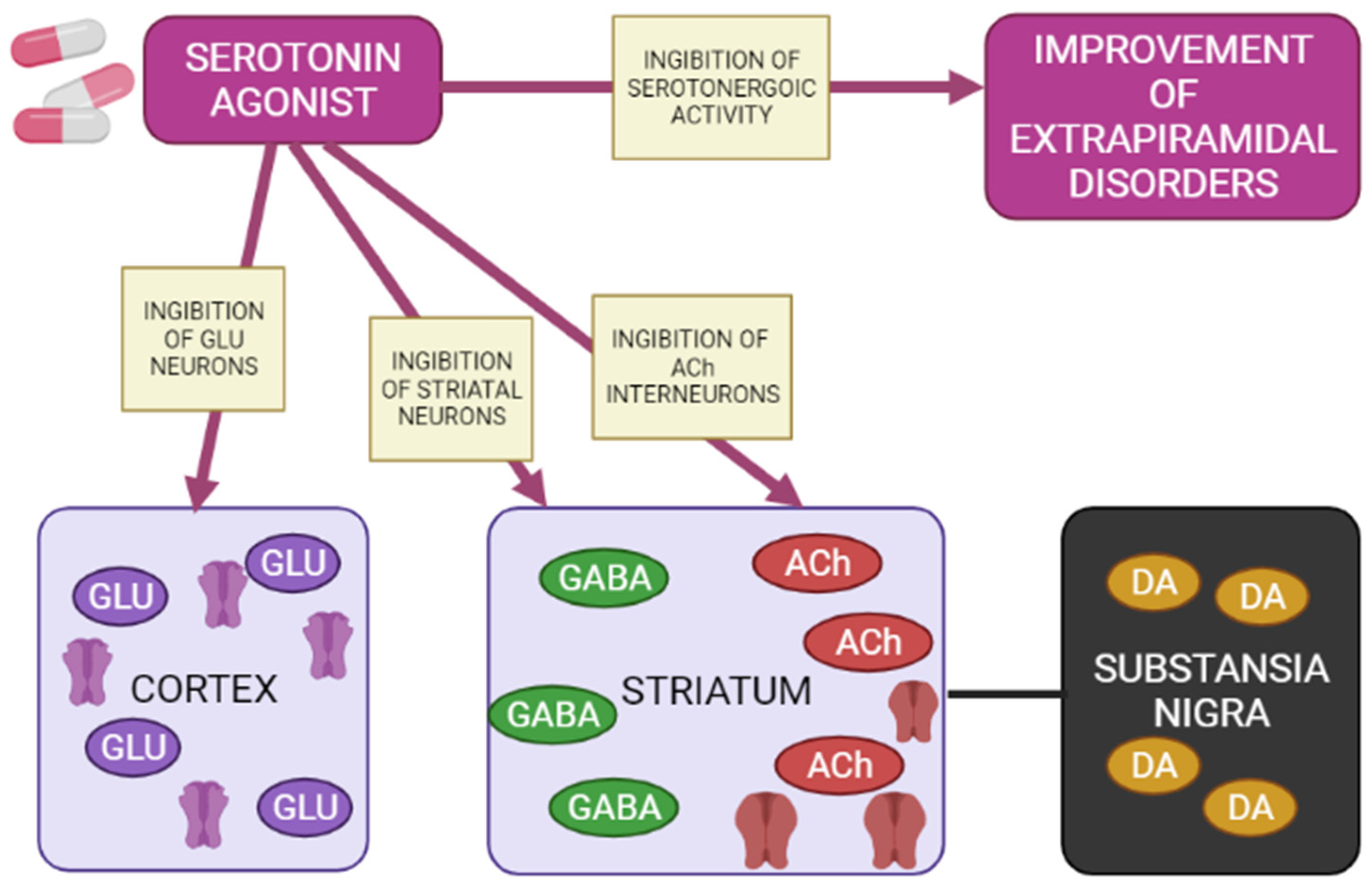
Agonists of postsynaptic and presynaptic serotonin 5-HT1A receptors lead to a de-crease in the manifestations of extrapyramidal movement disorders. This is explained by hyperpolarization GABA medial spine neurons or indirectly by inhibition acetylcholinergic and glutamatergic interneurons in the striatum. In addition, when presynaptic 5-HT1A autoreceptors are stimulated, the serotonergic activity of neurons in the nuclei of the sutures is inhibited, reducing the functions of 5-HT2A/2C, 5-HT3 and 5-HT6 receptors, as a result of which the symptoms of extrapyramidal movement disorders improve.
References
- Shin, H.W.; Chung, S.J. Drug-induced parkinsonism. J. Clin. Neurol. 2012, 8, 15–21.
- Nyberg, S.; Dencker, S.J.; Malm, U.; Dahl, M.-L.; Svetnson, J.-O.; Halldin, C.; Naskashima, Y.; Farde, L. D(2)- and 5-Ht(2) receptor occupancy in high-dose neuroleptictreated patients. Int. J. Neuropsychopharmacol. 1998, 1, 95–101.
- Crocker, A.D.; Hemsley, K.M. An animal model of extrapyramidal side effects induced by antipsychotic drugs: Relationship with D2 dopamine receptor occupancy. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 573–590.
- Farde, L.; Nordstrom, A.L.; Wiesel, F.A.; Pauli, S.; Halldin, C.; Sedvall, G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch. Gen. Psychiatry 1992, 49, 538–544.
- Scharrer, J.; Tatsch, K.; Schwarz, J.; Oertel, W.H.; Konjarczyk, M.; Albus, M. D2-dopamine receptor occupancy differs between patients with and without extrapyramidal side effects. Acta Psychiatr. Scand. 1994, 90, 266–268.
- Haddad, P.M.; Dursun, S.M. Neurological complications of psychiatric drugs: Clinical features and management. Hum. Psychopharmacol. 2008, 23 (Suppl. 1), S15–S26.
- Margolese, H.C.; Chouinard, G.; Kolivakis, T.T.; Beauclair, L.; Miller, R. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: Pathophysiology and mechanisms of induction. Can. J. Psychiatry. 2005, 50, 541–547.
- Sebastiao, A.M.; Ribeiro, J.A. Adenosine receptors and the central nervous system. Handb. Exp. Pharmacol. 2009, 193, 471–534.
- Grunder, G.; Carlsson, A.; Wong, D.F. Mechanism of new antipsychotic medications: Occupancy is not just antagonism. Arch. Gen. Psychiatry 2003, 60, 974–977.
- Sharma, A.; Sorrell, J.H. Aripiprazole-induced parkinsonism. Int. Clin. Psychopharmacol. 2006, 21, 127–129.
- Gunne, L.M.; Andrén, P.E. An animal model for coexisting tardive dyskinesia and tardive parkinsonism: A glutamate hypothesis for tardive dyskinesia. Clin. Neuropharmacol. 1993, 16, 90–95.
- Snyder, S.; Greenberg, D.; Yamamura, H.I. Antischizophrenic drugs and brain cholinergic receptors. Affinity for muscarinic sites predicts extrapyramidal effects. Arch. Gen. Psychiatry. 1974, 31, 58–61.
- Susatia, F.; Fernandez, H.H. Drug-induced parkinsonism. Curr. Treat. Options Neurol. 2009, 11, 162–169.
- Ward, K.M.; Citrome, L. Antipsychotic-Related Movement Disorders: Drug-Induced Parkinsonism vs. Tardive Dyskinesia-Key Differences in Pathophysiology and Clinical Management. Neurol. Ther. 2018, 7, 233–248.
- Vaiman, E.E.; Shnayder, N.A.; Neznanov, N.G.; Nasyrova, R.F. Pathophysiological mechanisms underlying antipsychotic-induced tardive dyskinesia. Bull. Sib. Med. 2019, 18, 169–184.
- Ossowska, K. Neuronal basis of neuroleptic-induced extrapyramidal side effects. Pol. J. Pharmacol. 2002, 54, 299–312.
- Powell, A.; Gallur, L.; Koopowitz, L.; Hayes, M.W. Parkinsonism in the psychiatric setting: An update on clinical differentiation and management. BMJ Neurol. Open 2020, 2, e000034.
- Kapur, S.; Seeman, P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. Am. J. Psychiatry 2001, 158, 360–369.
- Bohlega, S.A.; Al-Foghom, N.B. Drug-induced Parkinson’s disease. A clinical review. Neurosci. J. 2013, 18, 215–221.
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J Mol. Neurosci. 2005, 26, 209–220.
- Kulisevsky, J.; Poyurovsky, M. Adenosine A2A-receptor antagonism and pathophysiology of Parkinson’s disease and drug-induced movement disorders. Eur. Neurol. 2012, 67, 4–11.
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.; Chen, J. Adenosine, adenosine A 2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413.
- Hettinger, B.D.; Lee, A.; Linden, J.; Rosin, D.L. Ultrastructural localization of adenosine A2A receptors suggests multiple celular sites for modulation of GABAergic neurons in rat striatum. J. Comp. Neurol. 2001, 431, 331–346.
- Varty, G.B.; Hodgson, R.A.; Pond, A.J.; Grzelak, M.E.; Parker, E.M.; Hunter, J.C. The effects of adenosine A2A receptor antagonists on haloperidol-induced movement disorders in primates. Psychopharmacology 2008, 200, 393–401.
- Müller, C.E.; Ferre, S. Blocking striatal adenosine A 2A receptors: A new strategy for basal ganglia disorders. Recent Pat. CNS Drug Discov. 2007, 2, 1–21.
- Dayne, M.R.; Larson, G.; Orona, R.A.; Zahniser, N.R. Opposing actions of adenosine A2a and dopamine D2 receptor activation on GABA release in the basal ganglia: Evidence for an A2a/D2 receptor interaction in globus pallidus. Synapse 1996, 22, 132–138.
- Ferre, S.; Fredholm, B.B.; Morelli, M.; Popoli, P. Adenosine-dopamine receptor- receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487.
- Parsons, B.; Togasaki, D.M.; Kassir, S.; Przedborski, S. Neuroleptics up-regulate adenosine A 2a receptors in rat striatum: Im- 487 plications for the mechanism and the treatment of tardive dyskinesia. J. Neurochem. 1995, 65, 2057–2064.
- Bishnoi, M.; Chopra, K.; Kulkarni, S.K. Involvement of adenosinergic receptor system in an animal model of tardive dyskinesia and associated behavioural, biochemical and neurochemical changes. Eur. J. Pharmcol. 2006, 552, 55–66.
- Bishnoi, M.; Chopra, K.; Kulkarni, S.K. Theophylline, adenosine receptor antagonist prevents behavioral, biochemical and neurochemical changes associated with an animal model of tardive dyskinesia. Pharmacol. Rep. 2007, 59, 181–191.
- Wardas, J.; Konieczny, J.; Lorenc-Koci, E. SCH 58261, an A(2A) adenosine receptor antagonist, counteracts parkinsonian-like muscle rigidity in rats. Synapse 2001, 41, 160–171.
- John, D.S.; Salamone, J.D.; Betz, A.J.; Ishiwari, K.; Felsted, J.; Madson, L.; Mirante, B.; Clark, K.; Font, L.; Korbey, S.; et al. Tremorolytic effects of adenosine A2A antagonists: Implications for parkinsonism. Front. Biosci. 2008, 13, 3594–3605.
- Roth, B.L. Multiple serotonin receptors: Clinical and experimental aspects. Ann. Clin. Psychiatry. 1994, 6, 67–78.
- Baumgarten, H.G.; Grozdanovic, Z. Psychopharmacology of central serotonergic systems. Pharmacopsychiatry 1995, 28 (Suppl. 2), 73–79.
- Ohno, Y. Therapeutic role of 5-HT1A receptors in the treatment of schizophrenia and Parkinson’s disease. CNS Neurosci. Ther. 2011, 17, 58–65.
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152.
- Ohno, Y.; Tatara, A.; Shimizu, S.; Sasa, M. Management of Cognitive Impairments in Schizophrenia: The Therapeutic Role of 5-HT Receptors. In Schizophrenia Research: Recent Advances; Sumiyoshi, T., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; pp. 321–335.
- Ohno, Y.; Shimizu, S.; Imaki, J.; Masui, A.; Tatara, A. Management of Antipsychotic-Induced Extrapyramidal Motor Disorders: Regulatory Roles of the Serotonergic Nervous System. In Antipsychotic Drugs: Pharmacology, Side Effects and Abuse Prevention; Schwartz, T.L., Topel, M., Menga, J.L., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2013; pp. 219–234.
- Pucadyil, T.J.; Kalipatnapu, S.; Chattopadhyay, A. The serotonin1A receptor: A representative member of the serotonin receptor family. Cell. Mol. Neurobiol. 2005, 25, 553–580.
- Neal-Beliveau, B.S.; Joyce, J.N.; Lucki, I. Serotonergic involvement in haloperidol-induced catalepsy. J. Pharmacol. Exp. Ther. 1993, 265, 207–217.
- Wadenberg, M.L.; Young, K.A.; Richter, J.T.; Hicks, P.B. Effects of local application of 5-hydroxytryptamine into the dorsal or median raphe nuclei on haloperidol-induced catalepsy in the rat. Neuropharmacology 1999, 38, 151–156.
- Mignon, L.; Wolf, W.A. Postsynaptic 5-HT1A receptors mediate an increase in locomotor activity in the monoamine-depleted rat. Psychopharmacology 2002, 163, 85–94.
- Ohno, Y.; Shimizu, S.; Imaki, J. Evaluation of the antibradykinetic actions of 5-HT1A agonists using the mouse pole test. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1302–1307.
- Ohno, Y.; Shimizu, S.; Imaki, J. Anticataleptic 8-OH-DPAT preferentially counteracts with haloperidol-induced Fos expression in the dorsolateral striatum and the core region of the nucleus accumbens. Neuropharmacology 2008, 55, 717–723.
- Ohno, Y.; Shimizu, S.; Imaki, J. Effects of tandospirone, a 5-HT1A agonistic anxiolytic agent, on haloperidol-induced catalepsy and forebrain Fos expression in mice. J. Pharmacol. Sci. 2009, 109, 593–599.
- Shimizu, S.; Tatara, A.; Imaki, J.; Ohno, Y. Role of cortical and striatal 5-HT1A receptors in alleviating antipsychotic-induced extrapyramidal disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 877–881.
- Meltzer, H.Y. The mechanism of action of novel antipsychotic drugs. Schizophr. Bull. 1991, 17, 263–287.
- Ohno, Y.; Ishida-Tokuda, K.; Ishibashi, T. Potential role of 5-HT2 and D2 receptor interaction in the atypical antipsychotic action of the novel succimide derivative, perospirone. Pol. J. Pharmacol. 1997, 49, 213–219.
- Kapur, S.; Remington, G. Atypical antipsychotics: New directions and new challenges in the treatment of schizophrenia. Annu. Rev. Med. 2001, 52, 503–517.
- Livesey, M.R.; Cooper, M.A.; Deeb, T.Z. Structural determinants of Ca2+ permeability and conduction in the human 5-hydroxytryptamine type 3A receptor. J. Biol. Chem. 2008, 283, 19301–19313.
- Zhang, Z.J.; Kang, W.H.; Li, Q. Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: A double-blind, randomize, placebo-controlled study. Schizophr. Res. 2006, 88, 102–110.
- Akhondzadeh, S.; Mohammadi, N.; Noroozian, M.; Karamghadiri, N.; Ghoreishi, A.; Jamshidi, A.-H.; Forghani, S. Added ondansetron for stable schizophrenia: A double blind, placebo controlled trial. Schizophr. Res. 2009, 107, 206–212.
- Bonsi, P.; Cuomo, D.; Ding, J.; Sciamanna, G.; Ulrich, S.; Tscherter, A.; Bernardi, G.; Surmeier, D.J.; Pisani, A. Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT2C, 5-HT6, and 5-HT7 serotonin receptors: Implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology 2007, 32, 1840–1854.
- Ohno, Y.; Imaki, J.; Mae, Y. Serotonergic modulation of extrapyramidal motor disorders in mice and rats: Role of striatal 5-HT3 and 5-HT6 receptors. Neuropharmacology 2011, 60, 201–208.
- Tatara, A.; Shimizu, S.; Shin, N.; Sato, M.; Sugiuchi, T.; Imalki, J.; Ohno, Y. Modulation of antipsychotic-induced extrapyramidal side effects by medications for mood disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 38, 252–259.
- Ohno, Y.; Shimizu, S.; Tokudome, K. Pathophysiological roles of serotonergic system in regulating extrapyramidal motor functions. Biol. Pharm. Bull. 2013, 36, 1396–1400.
- Ebmeier, K.P.; O’Brien, J.T.; Taylor, J.-P. Psychiatry of Parkinson’s Disease. Adv. Biol. Psychiatry 2012, 12, 49–60.
More