Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Takumi Kitamoto and Version 2 by Camila Xu.
The discovery of the KCNJ5 somatic mutation in aldosteroene producing adenoma (APA) in 2011 and the development of specific CYP11B2 antibodies in 2012 have greatly advanced our understanding of the pathophysiology of primary aldosteronism. PA leads to abnormalities in the renin-angiotensin-aldosterone system and is known to increase the incidence of atrial fibrillation, heart failure, and stroke by 3.52, 2.05, and 2.58 times compared to hypertension due to essential hypertension.
- primary aldosteronism
- KCNJ5
- aldosterone producing adenoma
1. Introduction
Hypertension is the leading risk factor for death and disability in the global population [1][2][3][4][1,2,3,4]. The treatment goal of hypertensive patients is to maintain a healthy life expectancy comparable to that of healthy individuals. To achieve it, rwesearchers need to cure the disease and reduce the risk of cardiovascular complications as much as possible. In this regard, an appropriate classification of hypertension is necessary. Secondary hypertension should be screened for the patients showing following characteristics [5][6][7][5,6,7]: (1) onset of hypertension (<30 years), (2) abrupt onset of hypertension, (3) drug-resistant hypertension, (4) exacerbation of previously controlled hypertension, (5) suspicion of endocrine causes of hypertension or CKD, (6) clinical features suggestive of obstructive sleep apnoea, (7) unprovoked or excessive hypokalemia, (8) disproportionate targeted organ damage for degree of hypertension, (9) onset of diastolic hypertension in older adults (≥65 years). Primary aldosteronism (PA) is found in 5–7% of all hypertensive patients [8][9][10][11][12][13][8,9,10,11,12,13] and even explains 20% of patients with third-degree hypertension [14]. Despite its frequency, less than 1% of patients with PA undergo screening and progress to treatment throughout their lifetime [15][16][17][18][19][20][15,16,17,18,19,20]. One reason for the underdiagnosis may be that health care providers are not fully aware of the high frequency of PA in hypertensive patients. In addition, there is not sufficient consensus on the targets and methods of screening tests. In most of the institutions, patients to be screened are those with severe hypertension [21][22][23][21,22,23], i.e., in addition to the characteristics indicating secondary hypertension, (1) hypertension and spontaneous or diuretic-induced hypokalemia, (2) hypertension and adrenal incidentaloma, (3) atrial fibrillation in the absence of structural heart disease, (4) a family history of early onset hypertension or stroke at a young age (<40 years), (5) all hypertensive first-degree relatives of patients with PA. Others recommend screening for all hypertensive patients [24] from a cost-effectiveness perspective based on its high frequency [25][26][25,26] and the need for lifelong antihypertensive medication. With suspicions of PA at the first screening test evaluating plasma aldosterone levels and plasma renin activity, rwesearchers should move to definitive diagnosis of PA, performing confirmatory tests (Figure 1).
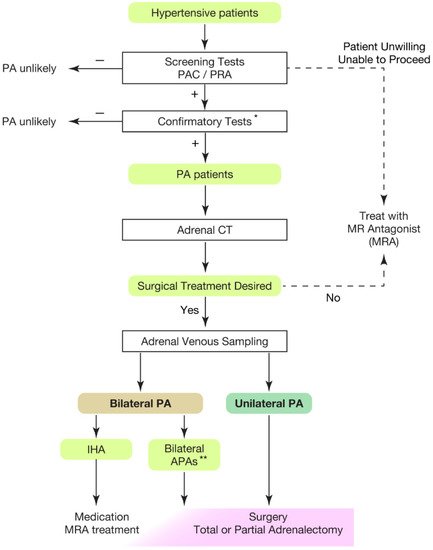
Figure 1. Overview of case detection and treatment strategy of primary aldosteronism. Most guidelines recommend to use plasma aldosterone-renin ratio (PAC/PRA) for screening test [21][22][23][24][27][21,22,23,24,33]. * Confirmatory tests included captopril challenge test, saline infusion test, and oral sodium loading test. ** Bilateral APAs can be diagnosed by segment selective adrenal venous sampling [28][29][34,35]. Partial adrenalectomy to spare normal adrenal tissue should be considered for the dominant side of APA if attempting surgical treatment. This would be helpful when the residual APA may cause MRA-resistant PA in the future. PAC, plasma aldosterone concentration; PRA, plasma renin activity; PA, primary aldosteronism; MRA, mineral corticoid receptor antagonist; CT, computed tomography; IHA, idiopathic hyperaldosteronism; APA, aldosterone producing adenoma.
2. Cardiovascular Outcome of Primary Aldosteronism
PA leads to abnormalities in the renin-angiotensin-aldosterone system and is known to increase the incidence of atrial fibrillation, heart failure, and stroke by 3.52, 2.05, and 2.58 times compared to hypertension due to essential hypertension [30][31][32][33][36,37,38,39]. Appropriate treatment for PA can greatly improve the prognosis of cardiovascular complications [32][38], and thus early diagnosis of PA and specific treatment for the condition are important. The etiology of PA is caused by two broad categories: aldosterone-producing adenomas (APA) and idiopathic hyper aldosteronism (IHA). In the former, the pathogenetic localization is clear, and surgical resection can be expected to cure the disease [34][40]. The frequency of APA and IHA has been reported to vary between primary care and referral centers [11][12][35][11,12,41], but it is difficult to determine the exact frequency. APA should be pathologically diagnosed by morphology and staining for CYP11B2 [36][42], an aldosterone-producing rate-limiting enzyme. However, surgical resection is only performed in cases who want to receive surgery and have been pre-surgically diagnosed with APA by adrenal venous sampling (AVS). IHA, on the other hand, is diagnosed by AVS and is usually treated with medication. We, therefore, cannot rule out the possibility of mixed misdiagnosis of APA in most cases. The frequency of APA and IHA diagnoses depends largely on the availability, procedure (e.g., usage of ACTH stimulation) and diagnostic criteria of AVS. Recently, tributary adrenal vein in addition to central adrenal vein sampling (a.k.a., segment selective AVS) has revealed that more than 10% of patients were elucidated as having APA, which could not be diagnosed by classic AVS evaluation of left-right difference [28][34]. Therefore, it is important to identify the localization of increased aldosterone synthesis within the adrenal gland to accurately diagnose APA, rather than simply looking at the right or left side of the adrenal gland. However, as a practical matter, the number of facilities where AVS is available is still far less than the expected number of patients with PA, and it is difficult to perform AVS on all patients with PA. Furthermore, publications reporting use of segment selective AVS are mostly from Japan [28][29][37][38][34,35,43,44]. Standardization and cost reduction of the AVS technique would give us precise etiology of APA and IHA. The data would help us to develop the diagnostic algorithm to predict the cases who require AVS.
3. Great Progress in Our Pathophysiological Understanding of PA
Along with the improvement of diagnostic techniques for PA, there have been remarkable advances in pathophysiological understanding of PA. One is the development of the specific CYP11B2 antibody, and the other is the findings of somatic mutations in APAs.
The corner stone of the specific monoclonal antibody against CYP11B2 was reported in 2014 [39][40][45,46]. CYP11B2 is the rate-limiting enzyme for aldosterone synthesis and the antibody was recently used for standardization for the nomenclature and criteria of the pathological diagnosis of surgical specimens of PA patients (HISTALDO) [36][42]. Classically, the diagnosis of aldosterone-producing adenoma and nodule was made morphologically by HE staining of the resected specimen (classic form). On the other hand, CYP11B2 staining has made it possible to diagnose multiple aldosterone-producing nodules or, less frequently, aldosterone-producing diffuse hyperplasia. In addition, this pathological diagnosis was reproducible among different pathologists (non-classic form). This standardization of the pathological diagnosis was built up by the incorporation of a functional aspect using CYP11B2 into the diagnosis, in parallel with the conventional morphological criteria. A prospective cohort study using these diagnostic concepts showed that non-classical multiple nodules were associated with more postoperative biochemical failures compared to patients diagnosed with a single macro lesion in its classic form represented by APA (33.3 vs. 2.4 (%)) [41][47]. The possibility that lesions with multiple nodules may be more prone to bilateral disease should be considered, although this is a matter of speculation due to the lack of pathological findings in the residual adrenal gland. Efforts should be made to make the preoperative diagnosis closer to the pathological diagnosis in order to take advantage of the finding that the two forms of classification would have different surgical implications for PA patients.
In parallel with these findings, the discovery of the aldosterone driver mutation has greatly advanced theour understanding of the pathogenesis of PA patients. The first step in this sequence was the memorable report published in 2011, describing a somatic mutation in the KCNJ5 gene encoding G protein-activated inward rectifier potassium channel 4 (GIRK-4) [42][48]. KCNJ5 mutation was identified in 36% of APA [42][43][44][48,49,50]. Following this discovery, somatic mutations in ATP2B3, ATP1A1 [45][51], and CACNA1D [46][52] were identified in 2013; although the frequency of KCNJ5 mutations in APA is higher in Eastern countries (59.5–76.8%) [47][48][49][50][53,54,55,56] than in Western countries (34–45%) [43][44][51][52][53][49,50,57,58,59], KCNJ5 mutations are similarly the most common mutation across the world. All of these mutations increase intracellular calcium concentrations, followed by activating CYP11B2 enzyme expression and increasing excess aldosterone biosynthesis. However, whether these mutations enhance cell proliferation has not been determined. Particularly, the proliferative effect seen in KCNJ5 was different due to KCNJ5 expression levels in the study using HAC15 [54][60], while another report using APAs did not show any evidence of proliferative effect of KCNJ5 mutation [55][61]. However, the evidence from germline KCNJ5 mutations suggested that KCNJ5 mutations promote proliferation to form hyperplasia in vivo [56][57][62,63]. A recent study using CYP11B2 immunohistochemistry-guided high-throughput sequencing rather than Sanger sequencing identified 95% of APAs as showing one of these somatic mutations, and more APAs with CACNA1D mutations among KCNJ5 wild-type APAs [58][64]. These mutations would play a crucial role in the pathogenesis of PA.
4. Pathological Insights into PA
CYP11B1 and CYCP11B2 play pivotal roles in the synthesis of adrenal corticosteroids, the former responsible for the synthesis of cortisol and corticosterone, and the latter for the synthesis of aldosterone [59][65]. The advent of CYP11B2 novel monoclonal antibodies has clarified the zonation of these synthesis enzymes in the normal adrenal gland [39][40][45,46]. Classically, the adrenal cortex is composed of three main zones, i.e., from the outermost layer, zona glomerulosa (ZG), to the zona fasciculata (ZF) and zona reticularis. Aldosterone and cortisol are produced by ZG and ZF cells, which have been shown by HE staining to be morphologically distinct: ZG cells are smaller, more compact, and have a smaller cytoplasm/nucleus ratio; ZF cells have a larger cytoplasm/nucleus ratio, with more fatty material and transparent appearance. Evaluation of the distribution of each synthase in the normal adrenal gland using specific CYP11B2 and B1 antibodies has shown that the distribution is different in young and elderly individuals [60][61][62][66,67,68]. In younger individuals, CYP11B2-positive cells are present throughout the ZG [61][67]; CYP11B1 was detected in both the ZF and zona reticularis. An unstained region is identified between the CYP11B2- and CYP11B1-positive cells that is not detected by either antibody. It is not clear whether this region comprises undifferentiated progenitor-like cells [63][69]. On the other hand, in the elderly, CYP11B2-positive cells are adjacent to the membrane and form a pattern of CYP11B2-immunopositive cell clusters with no CYP11B2 staining around them. Approximately half of the adrenals of normotensive individuals contain these regions, which have been designated as Aldosterone Producing Cell Clusters (APCCs) [60][61][62][64][65][66,67,68,70,71]. The nomenclature of APCC was changed to Aldosterone Producing Micronodules (APM) in the HISTALDO study [36][42]. The question of whether APMs have an autonomous aldosterone secretory capacity has been arisen. This is linked to the fact that the incidence of APMs correlates with the frequency of aldosterone-related hypertension [62][66][68,72]. Subsequent studies have shown that these APMs when found in adult human adrenal tissue with normal adrenal function also contain somatic mutations of the aldosterne driver mutation; mutations were identified in over 30% of APMs, most frequently in the CACNA1D mutation [58][60][65][64,66,71]. This was followed by ATP2B3, ATP1A1, and few KCNJ5 mutations. Interestingly, in case reports suggesting APM to APA transition, APM and micro APA were adjacent, with ATP1A1 and ATP2B3 mutations from the former and KCNJ5 mutations from the latter [67][68][73,74]. The research efforts have also extended to IHA cases. The study showed somatic mutations in CACNA1D are the cause of most IHA [69][75]. This is consistent with previous reports of image-negative PAs [70][76]. Interpretation is limited due to the small sample of surgically resected IHAs. However, their findings were much appreciated as supporting the concept of a continuum of pathophysiology from normotensive to hypertensive patients [71][77]. The different distribution of somatic mutations, with KCNJ5 mutations found specifically in APA and CACNA1D mutations in IHA, may contribute to the difference in clinical features between APA and IHA [35][41].
What is the pathological characteristics of APA harboring KCNJ5, and are they defined by the expression profile of steroidogenic enzymes? (See Figure 2a.) KCNJ5 mutated APAs are larger than other APAs; 60% are composed of lipid-laden clear ZF like cells and 40% are composed of compact ZG like cells [72][78]. Immunohistological studies on steroid synthase have shown that KCNJ5 mutated APAs are positive for CYP11B2 alone, co-expressing CYP11B2 and CYP17, and in a few cases, cells co-expressing CYP11B2, CYP11B1 and CYP17 [40][72][73][46,78,79]. On the other hand, APA with ATPA2B3 mutations tended to be dominated by compact ZG like cells, while APA with ATP1A1 and CACNA1D mutations showed heterogeneity from tumor to tumor with no clear advantage [49][72][55,78]. Very interestingly, the expression of CYP11B2 and CYP17A1, which are involved in aldosterone and cortisol synthesis, correlated positively in KCNJ5 mutant APAs, but negatively in ATP2B3 mutants [72][78]. The correlation between the two is not clear for ATP1A1 or CACNA1D. Inconsistent with this profile of steroidogenic enzymes, the normally negligible hybrid steroid, 18 oxocortisol (see the section “Development of prediction model for somatic KCNJ5 mutation in PA patinets”), is markedly increased in patients with APAs harboring KCNJ5 mutations. This increase has not been observed in patients with APAs harboring ATPase and CACNA1D mutations [58][74][64,80] (Figure 2a). This series of results has suggested that KCNJ5 mutations are associated with the formation of APA and that the pathogenetic process differs from that of ATPase and CACNA1D mutations. APAs harboring KCNJ5 mutations may have a more disorganized process of tumor cell differentiation and formation than APAs with ATPase or CACNA1D mutations. It has not been clear how the KCNJ5 mutation occurred in APAs.
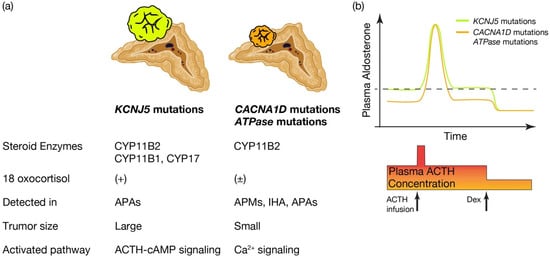
Figure 2. Distinctive characteristics of APA patients harboring KCNJ5 mutations, and CACNA1D or ATPase mutations. Distinctive characteristics of aldosterone producing adenoma (APA) patients harboring KCNJ5 mutation and CACNA1D or ATPase mutations. (a) KCNJ5 mutated APAs showed larger tumor and heterogenous composition of CYP11B2, CYP11B1, and/or CYP17 positive cells, while CACNA1D and ATPase mutated APAs showed smaller tumor and homogeneous composition of CYP11B2 positive cells. 18 oxocortisol is elevated in KCNJ5 mutated APAs, while not in CACNA1D and ATPase mutated APAs. KCNJ5 mutations have been detected mostly in APAs, while CACNA1D and ATPase mutations have been dominantly identified in aldosterone producing micronodules (APMs) and idiopathic hyperaldosteronism (IHA) as well as APAs. RWesearchers assume distinctive activated pathways of aldosterone synthesis between KCNJ5 mutated and CACNA1D or ATPase mutated APAs, such as the ACTH-cAMP and Ca2+ signaling pathways. (b) Conceptual scheme of responsiveness of aldosterone secretion to ACTH between APAs harboring KCNJ5 and CACNA1D or ATPase mutations is shown. Basal aldosterone secretion is higher and its responsiveness to ACTH stimulation is lower in KCNJ5 mutated APAs than CACNA1D and ATPase mutated APAs. ACTH depletion via dexamethasone (Dex) suppression decreased plasma aldosterone levels from KCNJ5 mutated APAs to that from KCNJ5 wild APAs [49][75][76][55,81,82]. Thus rwesearchers assume ACTH-cAMP signaling in KCNJ5 mutated APAs is activated to increase basal aldosterone secretion and to lessen response to extra ACTH stimulation. Figure was created with BioRender.com.