Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Alessandra Caligiuri.
Hepatic fibrosis is a scar formation process consisting in altered deposition of extracellular matrix. Progression of fibrosis can lead to impaired liver architecture and function, resulting in cirrhosis and organ failure. In the liver, due to its high regenerative ability, the extent of fibrosis regression and reversion to normal architecture is higher than in other tissues, even in advanced disease.
- liver fibrosis
- fibrosis regression
- myofibroblasts
- HSCs
- ECM degradation
- therapies
1. Introduction
Chronic liver diseases caused by different agents may result in hepatic fibrosis, characterized by a sequence of events leading to excessive deposition of collagen and other extracellular matrix proteins, scar formation and altered liver structure and function, potentially conducting to organ failure in cirrhosis [1,2][1][2]. Although in the past years the fibrogenic process was considered a unidirectional and irreversible phenomenon, in the last decades reversal of fibrosis, upon removal of the damaging agent(s), has been described in several tissues. In the liver, due to its regenerative ability, the extent of fibrosis regression and restitution towards normal architecture is higher than in other tissues, even in advanced disease. In recent years, several clinical observations and experimental studies have improved the mechanistic understanding of the fibrogenic process, providing information on the molecular mechanisms underlying reversal of liver fibrosis. Currently, as reviewed in some articles [3,4,5][3][4][5] the basis of fibrosis resolution can be recapitulated in the following major points:
- (1) Interruption or removal of detrimental agent(s) causing chronic hepatic injury [6];
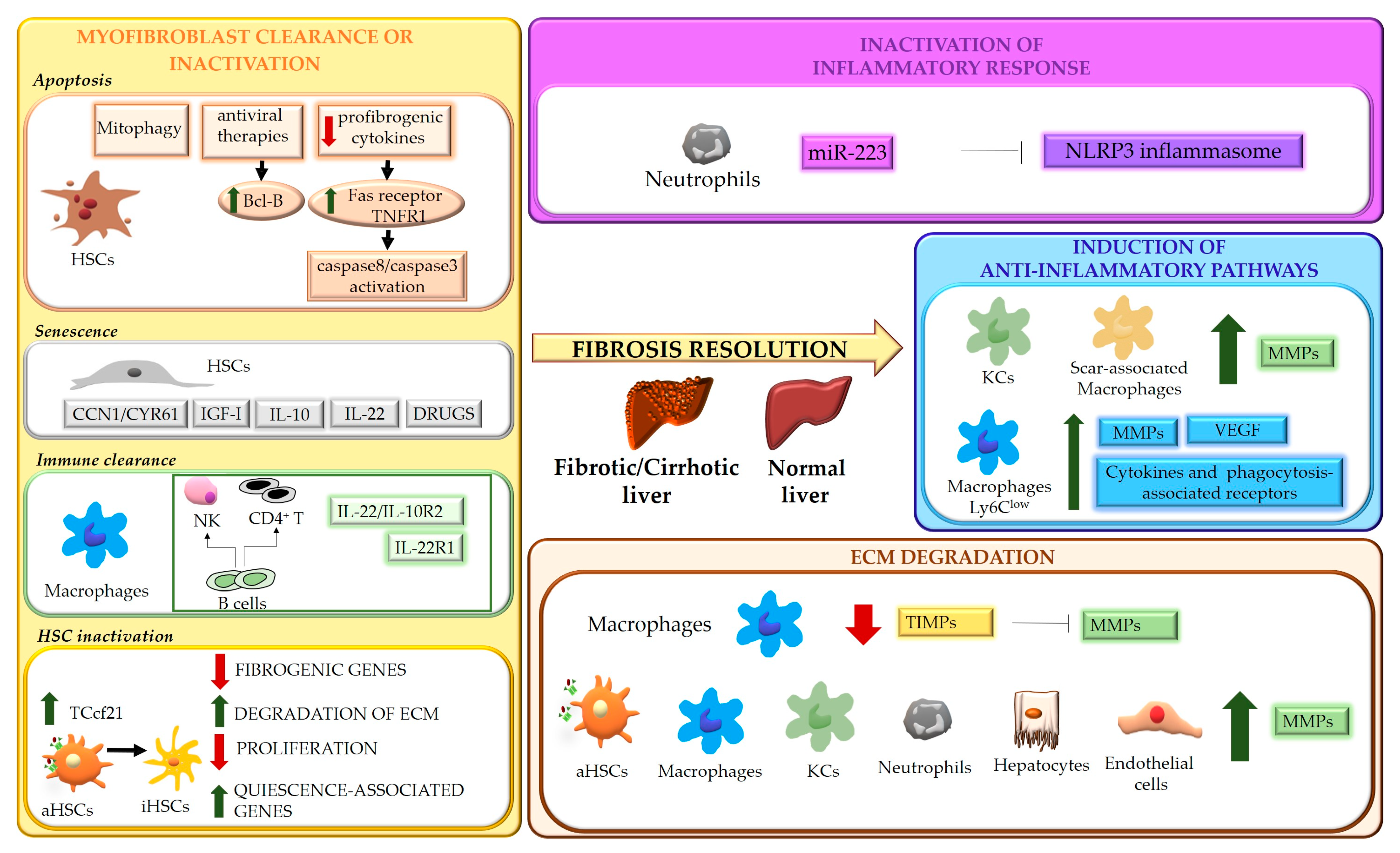
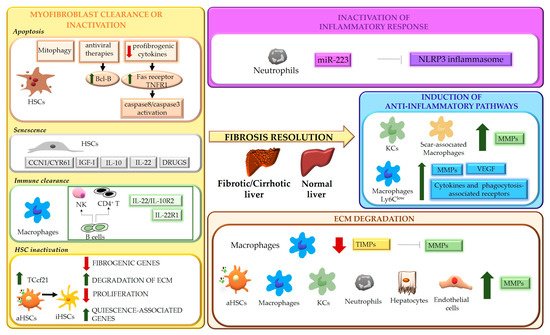
Figure 1. Schematic representation of the mechanisms underlying liver fibrosis regression. Four main mechanisms underlying the regression process of liver fibrosis are indicated. Hepatic stellate cells (HSCs); TNF receptor 1 (TNFR1); insulin-like growth factor I (IGF-I); transcription factor 21 (Tcf21); natural killer cells (NK); activated HSCs (aHSCs); inactivated HSCs (iHSCs); extracellular matrix (ECM); NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3); matrix metalloproteases (MMPs); Kupffer cells (KCs); vascular endothelial growth factor (VEGF); tissue inhibitors of MMPs (TIMPs).
2. Removal of Causative Agent(s)
Clinical evidence has recently demonstrated that compensated cirrhosis caused by chronic HBV or HCV infection is reversible following viral suppression or eradication [11,12][11][12]. These findings indicate that removal of the causative agent not only leads to interruption of fibrogenic signals, but also induces fibrolytic/restorative pathways, resulting in regression of fibrosis. However, a certain fraction of patients does not regress, suggesting a potential involvement of genetic/epigenetic mechanisms [13].
In experimental studies performed in mice treated with CCl4 to develop a pre-cirrhotic stage of liver injury and then allowed to spontaneously recover upon toxin withdrawal, resumption of CCl4 exposure rapidly induced profibrogenic features in HSCs, indicating that an “epigenetic memory” can be induced in these and, possibly, other cells [14,15][14][15].
3. Myofibroblast Clearance or Inactivation
The role of activated myofibroblasts in the development of liver fibrosis is well established. Different cell types can contribute to the myofibroblast population, including HSCs, portal fibroblasts, bone marrow-derived collagen producing cells (fibrocytes) and, possibly, parenchymal cells undergoing epithelial-mesenchymal transition (EMT) [49][16]. Although the origin of activated myofibroblasts may vary depending on the different etiologies of disease [50][17], HSCs can be considered their major source, as demonstrated by studies showing that HSCs depletion improves fibrosis in models based on both CCl4 intoxication and bile duct ligation [51][18]. Even in biliary fibrosis, where portal fibroblasts have been suggested to be the primary cell type initiating the fibrogenic response, giving rise to more than 70% of myofibroblasts, activation of HSCs becomes crucial after the initial phases [50][17]. During fibrosis regression, in response to a decrease of fibrogenic stimuli, the number of myofibroblasts drops, due to multiple mechanisms, that include restraint of activation, apoptosis, senescence, immune clearance, and reversal to a quiescent-like phenotype [14,15,52,53,54][14][15][19][20][21].4. Modulation of Inflammatory Processes
Inflammation represents a main feature of chronic liver diseases and plays a key role in any stages of the fibrogenic process, even during fibrosis regression. Inflammatory response involves multicellular interactions, dynamically regulated by a plethora of factors (e.g., soluble mediators, ECM components, pathogen-associated molecular patterns-PAMPs, damage-associated molecular patterns-DAMPs), acting in cell-specific fashion and aimed to restore liver architecture and function, but also leading to liver fibrosis when the noxious agent persists. Cell death is an early and primary inducer of chronic inflammation and fibrosis. Hepatocyte-derived apoptotic bodies stimulate the secretion of pro-inflammatory and profibrogenic cytokines from macrophages and promote activation of HSCs through induction of autophagy [100,101,102][22][23][24]. In addition, injured hepatocytes release DAMPs, such as ATP, phormyl peptides, High Mobility Group Box 1(HMGB1) [103][25] and cytokines such as IL-33 [104][26], which triggers HSC activation directly or indirectly, by promoting IL-13 release by innate lymphoid cells (ILC2). At the same time, inflammatory mediators secreted by infiltrating immune cells contribute to cell death, amplifying hepatic injury [104][26]. As major effectors of fibrosis, activated HSCs play a central role in inflammation, receiving a wide variety of stimuli from inflammatory cells and from hepatocytes, cholangiocytes and activated sinusoidal endothelial cells (SECs). Activated HSCs are highly responsive to inflammatory mediators which induce inflammatory pathways (such as NF-κB and AP-1) [105,106][27][28] and consequent secretion of cytokine/chemokines that act in autocrine and paracrine fashion. Inflammatory signals exert specific roles on HSCs, maintaining survival (IL-1β, TNFα, CXCL12) and the activated state (ILs and chemokines) [107][29], providing chemotactic stimuli for HSCs themselves or inflammatory cells (CCL2, CCL5, CXCL9, CXCL10, CX3CL1) and mediating the gut-liver axis crosstalk (toll like receptors (TLRs)) [105][27]. All these processes can contribute to positively or negatively modulate inflammatory responses and fibrogenesis, promoting fibrosis progression or regression. As modulators of liver fibrosis, immune cells exhibit a dual role, being able to contribute to both fibrosis progression and regression [108,109][30][31]. Danger signals generated in the site of injury lead to infiltration of circulating inflammatory cells (T lymphocytes, neutrophils, dendritic cells and monocytes) and activation of Kupffer cells (KCs) [108,109][30][31]. The release of a wide range of soluble mediators amplifies inflammation and stimulates the fibrogenic process. Upon removal of the cause of injury, the balance switches from pro- to anti-inflammatory/restorative pathways, promoting fibrosis resolution. This shift is achieved by rearrangements in the type of immune cell populations recruited, with a marked drop in intrahepatic T cells and blood-derived cells (NKT cells, monocytes) [110][32], and phenotypic modifications of certain cell types, mainly macrophages.5. ECM Degradation
Liver fibrosis is a dynamic process characterized by an unfavorable balance between ECM deposition and degradation. Degradation of ECM represents one of the most relevant aspects of fibrosis regression and requires activation of MMPs, macrophage phagocytic activity and downregulation of MMP-inhibitory molecules, such as tissue inhibitors of MMPs, TIMPs [7,10][7][10]. MMPs are the main matrix-degrading enzymes [62][33] and, according to substrate specificity, can be grouped in collagenases (MMP8, MMP1 and MMP13) which cleave native fibrillar collagens to gelatin, gelatinases (MMP2, MMP9), degrading a wide range of substrates including gelatin, collagens and, in some extent, elastin, metalloelastases (MMP12) and others (Table 1).Table 1.
Classification of human metalloproteinases (MMPs) and their function.
|
MMPs |
GROUP |
FUNCTION |
|---|---|---|
|
MMP1, MMP8, MMP13 |
Collagenases |
Cleavage of native fibrillar collagens to gelatin |
|
MMP2, MMP9 |
Gelatinases |
Degradation of a wide range of substrates, including gelatin, collagens and elastin |
|
MMP12 |
Metalloelastases |
Elastin degradation |
6. Reversibility of Cirrhosis
ECM remodeling is crucial in determining reversibility of fibrosis. In recent years, clinical and experimental studies have provided evidence that matrix remodeling and at least partial restitution towards a normal architecture may be observed even in advanced liver fibrosis or cirrhosis [136,137,138,139,140][36][37][38][39][40]. The amount of elastin and cross-linked proteins in fibrotic scars is critical in this process. Protein cross-linking, which is mediated by cellular transglutaminases and lysyl oxidases, stabilizes ECM, enhancing its resistance to enzyme degradation and, together with elastin, increases matrix stiffness, that further sustains HSC activation via integrin-mediated mechanisms [62][33]. In this setting, MMP12 released by macrophages can still promote matrix turnover acting not only on elastin but also on collagens [141][41]. However, ECM remodeling in cirrhotic scars is also influenced by vascular remodeling that can hamper matrix degradation [142][42]. Thus, even when restitution to normal liver architecture is achieved, cirrhosis-associated derangements in the vascular system and in other organs persist. By using two different models of cirrhosis induction and reversal (TAA and BDL), Hsu et al. demonstrated that, despite a complete regression of fibrotic scars, portal hypertension was only partially reduced, due to persisting alterations in splanchnic and collateral circulation [143][43]. These biologic considerations have clear clinical implications. Regression of fibrosis represents a major clinical goal, since it can lead to a recovery of liver function and reduction in portal pressure, which decrease the incidence of portal-hypertensive complications and of hepatocellular carcinoma (HCC) [144,145,146,147][44][45][46][47]. It is well known that mild and moderate fibrosis can be reversible, but the same concept is not always true for cirrhosis. In this respect, the identification of a “point of no return” in the natural history of liver disease can be very difficult, despite its utmost relevance in clinical practice. This may be viewed as a condition beyond which even causal therapy (e.g., viral eradication) does not determine a significant regression of fibrosis and/or has limited impact on the appearance of complications and prognosis of the patient. As indicated above, the degree and amount of structural damage, in particular the development of extensive matrix crosslinking [148][48] and accumulation of elastin fibers in long-standing cirrhosis, have been indicated as a major element to identify the “point of no return” [141][41]. From a clinical standpoint, the ‘model’ of HCV eradication has provided relevant data in this context. Patients with compensated cirrhosis (Child-Turcotte-Pugh class A) achieving viral eradication with direct-acting antivirals (DAAs), show regression of fibrosis in a relevant percentage of cases (88%) [149][49] and a consequent decrease of portal hypertension [150][50]. When patients with cirrhosis Child-Pugh class B and C are considered, long-term data about the effects of sustained virologic response (SVR) after DAA treatment on fibrosis and liver-related complications and survival are less abundant. However, data from other contexts (e.g., HBV or alcohol-related decompensated cirrhosis) indicated that Child C class could represent a “point of no return” in terms of fibrosis decrease even after removal of the etiologic factor [151,152][51][52]. Other clinical predictors of the lack of fibrosis regression include age (>65 years), albumin (<3.5 g/dL), high MELD score (>20), alcohol habit and presence of metabolic disorders. However, none of them are satisfactory consistent to be used in clinical practice [153][53]. Advanced liver fibrosis and cirrhosis are major risk factors for HCC [154,155][54][55]. In particular, fibrosis and cancer-associated fibroblasts (CAF,) can influence the onset of HCC modulating the cancer microenvironment [156,157][56][57]. Considering these assumptions, HCV eradication should determine a decrease of both HCC occurrence and recurrence. In recent years, this has been a very debated issue since some studies suggested that SVR due to DAA, differently from interferon-based therapies, could increase the risk of both occurrence and recurrence of liver cancer [158][58]. It is now accepted that there is no such risk on a population basis and, as recently demonstrated [159][59], SVR due to DAAs leads to a drop in all-cause mortality, hepatic decompensation, and HCC. Nevertheless, on an individual basis, DAAs might favor the HCC development in subjects who already have a predisposing hepatic condition such as activated neo angiogenesis [160][60]. Moreover, subjects with severe metabolic impairment may have a risk of HCC despite viral eradication [161][61]. DAA-induced modifications in VEGF, epidermal growth factor, and inflammatory factors have been proposed for the detection of subgroups at risk of HCC, and some authors have proposed these as possible determinants of the susceptibility to cancer development [162,163][62][63].7. Vascular Remodeling
As anticipated above, angiogenesis and vascular remodeling represent additional mechanisms involved in both fibrosis development and regression. Although the role of angiogenesis in promoting liver fibrosis is fully accepted, new lines of evidence indicate that angiogenic factors may also induce scar degradation and tissue repair during fibrosis resolution. Using murine models of fibrosis reversal, Yang et al. showed that the VEGF/VEGFR2 pathway is essential to maintaining sinusoidal permeability and the subsequent monocyte infiltration and macrophage fibrinolytic activity [128][64]. Moreover, VEGF release by macrophages was shown to be critical for fibrosis resolution. In fact, VEGR2-mediated activation by VEGF induced ECM degradation through upregulation of MMPs and downregulation of TIMPs in sinusoidal endothelial cells [164][65]. Capillarization of the sinusoids and changes in liver sinusoidal endothelial cells (LSECs) represent key events in liver fibrogenesis, triggering HSC activation and impairing hepatocyte polarization. These consist in LSEC dedifferentiation with loss of fenestrae and deposition of a continuous basement membrane that hampers normal exchanges between blood circulation and hepatocytes. Restoration of differentiated LSEC is crucial for recovery from hepatic fibrosis, as proved by the fact that depleting factors implicated in sinusoidal permeability, such as VEGF or CXCL9, results in delayed recovery [128][64]. In a thioacetamide-induced rat model of cirrhosis, administration of BAY 60-2770, an activator of soluble guanylate cyclase (sGC), promoted a complete reversal of sinusoid capillarization, by restoring normal levels of cGMP, fenestrae, and porosity in LSECs. Restitution to differentiated LSECs resulted in reversal of HSC activation and regression of fibrosis. Moreover, maintenance of physiological levels of cGMP in LSECs was essential to prevent fibrosis progression [165][66]. Liver X Receptor (LXR) α, which mediates multiple antifibrogenic actions interfering with the activation of HSCs, the release of inflammatory mediators and the synthesis of profibrogenic factors [56,166,167[67][68][69][70],168], was hypothesized to play a role in reverting capillarization of the sinusoids, through inhibition of Hedgehog-dependent signaling in LSECs [169][71]. In a mouse model of biliary fibrosis induction and reversal, Lee et al. identified AKAP12, a scaffold protein expressed in various cell types regulating cyclic adenosine monophosphate (cAMP) compartmentalization, as a novel mediator of fibrosis resolution, through mechanisms affecting LSEC dedifferentiation/activation and angiogenesis [170][72]. In an elegant study, Xu et al. identified leukocyte cell-derived chemotaxin 2 (LECT2) as a ligand of Tie1 (an orphan receptor expressed by endothelial cells) and LECT2-Tie1 as a novel profibrogenic pathway involved in vascular remodeling, that enhances sinusoid capillarization and reduces portal angiogenesis. They showed that knockdown of LECT2 (in both LECT2 KO mice and AAV9-LECT2 shRNA- treated mice) attenuates fibrosis development and ameliorates established fibrosis in different experimental models, reducing sinusoid capillarization and increasing portal angiogenesis. Notably, serum levels of LECT2 were significantly increased in patients with advanced fibrosis, indicating LECT2-Tie1 signaling as a promising therapeutic target [171][73].References
- Pinzani, M.; Rombouts, K.; Colagrande, S. Fibrosis in chronic liver diseases: Diagnosis and management. J. Hepatol. 2005, 42 (Suppl. 1), S22–S36.
- Khurana, A.; Sayed, N.; Allawadhi, P.; Weiskirchen, R. It’s all about the spaces between cells: Role of extracellular matrix in liver fibrosis. Ann. Transl. Med. 2021, 9, 728.
- Zoubek, M.E.; Trautwein, C.; Strnad, P. Reversal of liver fibrosis: From fiction to reality. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 129–141.
- Tacke, F.; Trautwein, C. Mechanisms of liver fibrosis resolution. J. Hepatol. 2015, 63, 1038–1039.
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15.
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96.
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107.
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283.
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76.
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56.
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475.
- D’Ambrosio, R.; Aghemo, A.; Rumi, M.G.; Ronchi, G.; Donato, M.F.; Paradis, V.; Colombo, M.; Bedossa, P. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology 2012, 56, 532–543.
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250.
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453.
- Troeger, J.S.; Mederacke, I.; Gwak, G.Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083.
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 65–68.
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305.
- Puche, J.E.; Lee, Y.A.; Jiao, J.; Aloman, C.; Fiel, M.I.; Muñoz, U.; Kraus, T.; Lee, T.; Yee, H.F.; Friedman, S.L. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 2013, 57, 339–350.
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667.
- Iredale, J.P.; Benyon, R.C.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M.J. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Investig. 1998, 102, 538–549.
- Kong, D.; Zhang, F.; Zhang, Z.; Lu, Y.; Zheng, S. Clearance of activated stellate cells for hepatic fibrosis regression: Molecular basis and translational potential. Biomed. Pharmacother. 2013, 67, 246–250.
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Investig. 2003, 83, 655–663.
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198.
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443.
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172.
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371.
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332.
- Hellerbrand, C.; Jobin, C.; Licato, L.L.; Sartor, R.B.; Brenner, D.A. Cytokines induce NF-kappaB in activated but not in quiescent rat hepatic stellate cells. Am. J. Physiol. 1998, 275, G269–G278.
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172.
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594.
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218.
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195.
- Feng, M.; Ding, J.; Wang, M.; Zhang, J.; Zhu, X.; Guan, W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int. J. Biol. Sci. 2018, 14, 1033–1040.
- Hammel, P.; Couvelard, A.; O’Toole, D.; Ratouis, A.; Sauvanet, A.; Fléjou, J.F.; Degott, C.; Belghiti, J.; Bernades, P.; Valla, D.; et al. Regression of liver fibrosis after biliary drainage in patients with chronic pancreatitis and stenosis of the common bile duct. N. Engl. J. Med. 2001, 344, 418–423.
- Desmet, V.J.; Roskams, T. Cirrhosis reversal: A duel between dogma and myth. J. Hepatol. 2004, 40, 860–867.
- Dienstag, J.L.; Goldin, R.D.; Heathcote, E.J.; Hann, H.W.; Woessner, M.; Stephenson, S.L.; Gardner, S.; Gray, D.F.; Schiff, E.R. Histological outcome during long-term lamivudine therapy. Gastroenterology 2003, 124, 105–117.
- Michalopoulos, G.K.; DeFrances, M. Liver regeneration. Adv. Biochem. Eng. Biotechnol. 2005, 93, 101–134.
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55.
- Pellicoro, A.; Aucott, R.L.; Ramachandran, P.; Robson, A.J.; Fallowfield, J.A.; Snowdon, V.K.; Hartland, S.N.; Vernon, M.; Duffield, J.S.; Benyon, R.C.; et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 2012, 55, 1965–1975.
- Wanless, I.R.; Nakashima, E.; Sherman, M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch. Pathol. Lab. Med. 2000, 124, 1599–1607.
- Hsu, S.J.; Tsai, M.H.; Chang, C.C.; Hsieh, Y.H.; Huang, H.C.; Lee, F.Y.; Chuang, C.L.; Hou, M.C.; Lee, S.D. Extrahepatic angiogenesis hinders recovery of portal hypertension and collaterals in rats with cirrhosis resolution. Clin. Sci. 2018, 132, 669–683.
- Calvaruso, V.; Craxì, A. Hepatic benefits of HCV cure. J. Hepatol. 2020, 73, 1548–1556.
- Van der Meer, A.J.; Berenguer, M. Reversion of disease manifestations after HCV eradication. J. Hepatol. 2016, 65 (Suppl. 1), S95–S108.
- Martini, S.; Sacco, M.; Strona, S.; Arese, D.; Tandoi, F.; Dell Olio, D.; Stradella, D.; Cocchis, D.; Mirabella, S.; Rizza, G.; et al. Impact of viral eradication with sofosbuvir-based therapy on the outcome of post-transplant hepatitis C with severe fibrosis. Liver Int. 2016, 37, 62–70.
- Pietsch, V.; Deterding, K.; Attia, D.; Ringe, K.I.; Heidrich, B.; Cornberg, M.; Gebel, M.; Manns, M.P.; Wedemeyer, H.; Potthoff, A. Long-term changes in liver elasticity in hepatitis C virus-infected patients with sustained virologic response after treatment with direct-acting antivirals. United Eur. Gastroenterol. J. 2018, 6, 1188–1198.
- Brenner, D.A. Reversibility of liver fibrosis. Gastroenterol. Hepatol. 2013, 9, 737–739.
- Knop, V.; Hoppe, D.; Welzel, T.; Vermehren, J.; Herrmann, E.; Vermehren, A.; Friedrich-Rust, M.; Sarrazin, C.; Zeuzem, S.; Welker, M.W. Regression of fibrosis and portal hypertension in HCV-associated cirrhosis and sustained virologic response after interferon-free antiviral therapy. J. Viral Hepat. 2016, 23, 994–1002.
- Mandorfer, M.; Kozbial, K.; Schwabl, P.; Freissmuth, C.; Schwarzer, R.; Stern, R.; Chromy, D.; Stättermayer, A.F.; Reiberger, T.; Beinhardt, S.; et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J. Hepatol. 2016, 65, 692–699.
- Fontana, R.J.; Hann, H.W.; Perrillo, R.P.; Vierling, J.M.; Wright, T.; Rakela, J.; Anschuetz, G.; Davis, R.; Gardner, S.D.; Brown, N.A. Determinants of early mortality in patients with decompensated chronic hepatitis B treated with antiviral therapy. Gastroenterology 2002, 123, 719–727.
- Vanlemmens, C.; Di Martino, V.; Milan, C.; Messner, M.; Minello, A.; Duvoux, C.; Poynard, T.; Perarnau, J.M.; Piquet, M.A.; Pageaux, G.P.; et al. Immediate listing for liver transplantation versus standard care for Child-Pugh stage B alcoholic cirrhosis: A randomized trial. Ann. Intern. Med. 2009, 150, 153–161.
- Vinaixa, C.; Strasser, S.I.; Berenguer, M. Disease reversibility in patients with post-hepatitis C cirrhosis: Is the point of no return the same before and after liver transplantation? A review. Transplantation 2017, 101, 916–923.
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127 (Suppl. 1), S35–S50.
- O’Rourke, J.M.; Sagar, V.M.; Shah, T.; Shetty, S. Carcinogenesis on the background of liver fibrosis: Implications for the management of hepatocellular cancer. World J. Gastroenterol. 2018, 24, 4436–4447.
- Affo, S.; Yu, L.X.; Schwabe, R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017, 12, 153–186.
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1723.
- Rutledge, S.M.; Zheng, H.; Li, D.K.; Chung, R.T. No evidence for higher rates of hepatocellular carcinoma after direct-acting antiviral treatment: A meta-analysis. Hepatoma Res. 2019, 5, 1–12.
- Ghany, M.G.; Morgan, T.R.; Panel, A.H.C. G Hepatitis C Guidance 2019 Update: American Association for the Study of Liver Diseases-Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology 2019, 71, 686–721.
- Faillaci, F.; Marzi, L.; Critelli, R.; Milosa, F.; Schepis, F.; Turola, E.; Andreani, S.; Vandelli, G.; Bernabucci, V.; Lei, B.; et al. Liver angiopoietin-2 is a key predictor of de novo or recurrent hepatocellular cancer after hepatitis C virus direct-acting antivirals. Hepatology 2018, 68, 1010–1024.
- Nahon, P.; Layese, R.; Bourcier, V.; Cagnot, C.; Marcellin, P.; Guyader, D.; Pol, S.; Larrey, D.; De Lédinghen, V.; Ouzan, D.; et al. Incidence of hepatocellular carcinoma after direct antiviral therapy for HCV in patients with cirrhosis included in surveillance programs. Gastroenterology 2018, 155, 1436–1450.
- Villani, R.; Facciorusso, A.; Bellanti, F.; Tamborra, R.; Piscazzi, A.; Landriscina, M.; Vendemiale, G.; Serviddio, G. DAAs rapidly reduce inflammation but increase serum VEGF level: A rationale for tumor risk during anti-HCV treatment. PLoS ONE 2016, 11, e0167934.
- Gardini, A.C.; Foschi, F.G.; Conti, F.; Petracci, E.; Vukotic, R.; Marisi, G.; Buonfiglioli, F.; Vitale, G.; Ravaioli, F.; Gitto, S.; et al. Immune inflammation indicators and ALBI score to predict liver cancer in HCV-patients treated with direct-acting antivirals. Dig. Liver Dis. 2019, 51, 681–688.
- Yang, L.; Kwon, J.; Popov, Y.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology 2014, 146, 1339–1350.
- Kantari-Mimoun, C.; Castells, M.; Klose, R.; Meinecke, A.K.; Lemberger, U.J.; Rautou, P.E.; Pinot-Roussel, H.; Badoual, C.; Schrödter, K.; Österreicher, C.H.; et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology 2015, 61, 2042–2055.
- Xie, G.; Wang, X.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918–927.
- Mallat, A.; Lotersztajn, S. The liver X receptor in hepatic stellate cells: A novel antifibrogenic target? J. Hepatol. 2011, 55, 1452–1454.
- Beaven, S.W.; Wroblewski, K.; Wang, J.; Hong, C.; Bensinger, S.; Tsukamoto, H.; Tontonoz, P. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology 2011, 140, 1052–1062.
- Wang, Y.Y.; Dahle, M.K.; Agren, J.; Myhre, A.E.; Reinholt, F.P.; Foster, S.J.; Collins, J.L.; Thiemermann, C.; Aasen, A.O.; Wang, J.E. Activation of the liver X receptor protects against hepatic injury in endotoxemia by suppressing Kupffer cell activation. Shock 2006, 25, 141–146.
- Königshofer, P.; Brusilovskaya, K.; Petrenko, O.; Hofer, B.S.; Schwabl, P.; Trauner, M.; Reiberger, T. Nuclear receptors in liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166235.
- Xing, Y.; Zhao, T.; Gao, X.; Wu, Y. Liver X receptor α is essential for the capillarization of liver sinusoidal endothelial cells in liver injury. Sci. Rep. 2016, 6, 21309.
- Lee, H.S.; Choi, J.; Son, T.; Wee, H.J.; Bae, S.J.; Seo, J.H.; Park, J.H.; Ryu, S.H.; Lee, D.; Jang, M.K.; et al. Altered AKAP12 expression in portal fibroblasts and liver sinusoids mediates transition from hepatic fibrogenesis to fibrosis resolution. Exp. Mol. Med. 2018, 50, 1–13.
- Xu, M.; Xu, H.H.; Lin, Y.; Sun, X.; Wang, L.J.; Fang, Z.P.; Su, X.H.; Liang, X.J.; Hu, Y.; Liu, Z.M.; et al. LECT2, a ligand for Tie1, plays a crucial role in liver fibrogenesis. Cell 2019, 178, 1478–1492.
More