Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alessandra Caligiuri and Version 2 by Peter Tang.
Hepatic fibrosis is a scar formation process consisting in altered deposition of extracellular matrix. Progression of fibrosis can lead to impaired liver architecture and function, resulting in cirrhosis and organ failure. In the liver, due to its high regenerative ability, the extent of fibrosis regression and reversion to normal architecture is higher than in other tissues, even in advanced disease.
- liver fibrosis
- fibrosis regression
- myofibroblasts
- HSCs
- ECM degradation
- therapies
1. Introduction
Chronic liver diseases caused by different agents may result in hepatic fibrosis, characterized by a sequence of events leading to excessive deposition of collagen and other extracellular matrix proteins, scar formation and altered liver structure and function, potentially conducting to organ failure in cirrhosis [1][2][1,2]. Although in the past years the fibrogenic process was considered a unidirectional and irreversible phenomenon, in the last decades reversal of fibrosis, upon removal of the damaging agent(s), has been described in several tissues. In the liver, due to its regenerative ability, the extent of fibrosis regression and restitution towards normal architecture is higher than in other tissues, even in advanced disease. In recent years, several clinical observations and experimental studies have improved the mechanistic understanding of the fibrogenic process, providing information on the molecular mechanisms underlying reversal of liver fibrosis. Currently, as reviewed in some articles [3][4][5][3,4,5] the basis of fibrosis resolution can be recapitulated in the following major points:
- (1) Interruption or removal of detrimental agent(s) causing chronic hepatic injury [6];
The mechanisms underlying the regression of fibrosis are summarized in Figure 1.
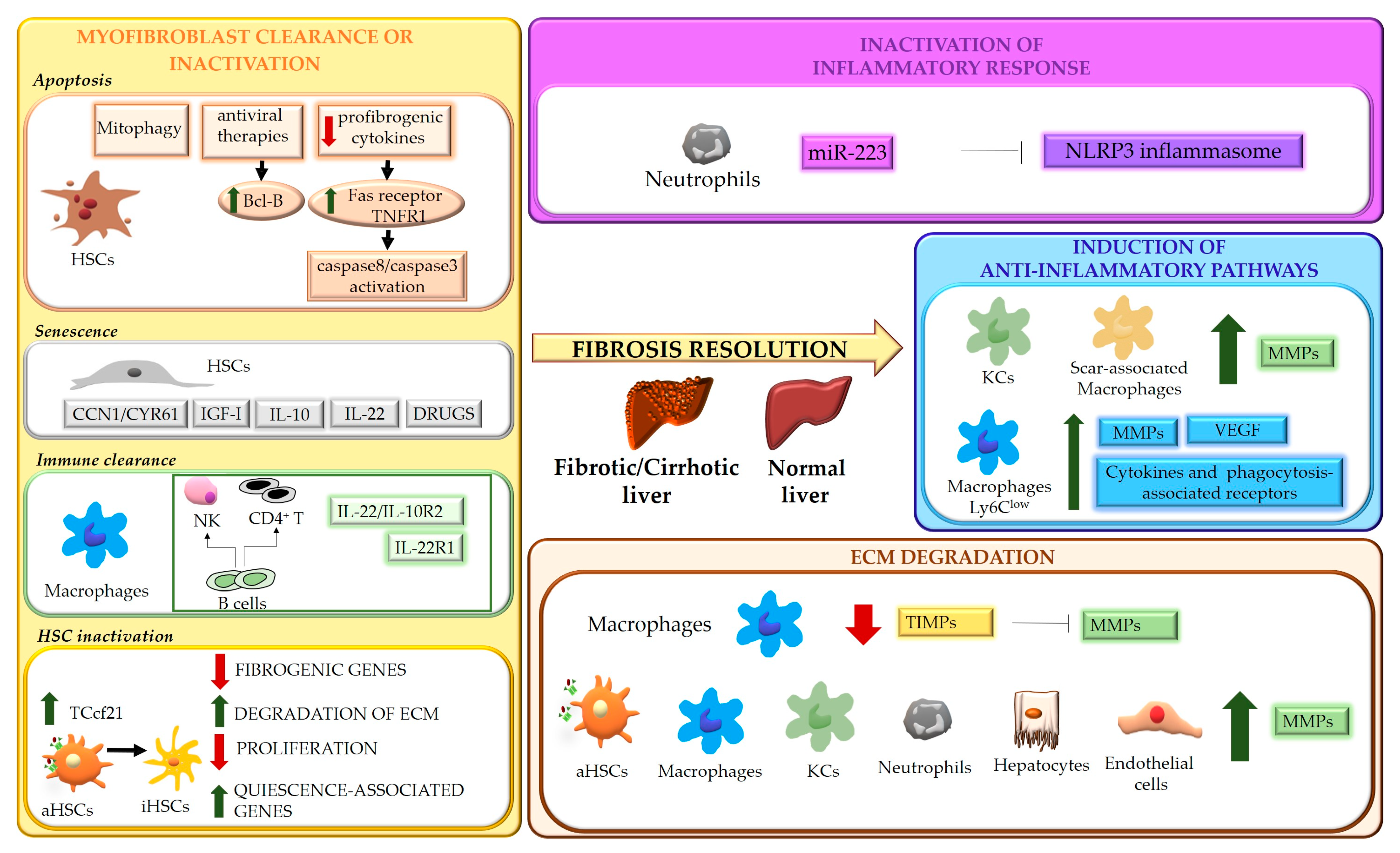
Figure 1. Schematic representation of the mechanisms underlying liver fibrosis regression. Four main mechanisms underlying the regression process of liver fibrosis are indicated. Hepatic stellate cells (HSCs); TNF receptor 1 (TNFR1); insulin-like growth factor I (IGF-I); transcription factor 21 (Tcf21); natural killer cells (NK); activated HSCs (aHSCs); inactivated HSCs (iHSCs); extracellular matrix (ECM); NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3); matrix metalloproteases (MMPs); Kupffer cells (KCs); vascular endothelial growth factor (VEGF); tissue inhibitors of MMPs (TIMPs).
2. Removal of Causative Agent(s)
Clinical evidence has recently demonstrated that compensated cirrhosis caused by chronic HBV or HCV infection is reversible following viral suppression or eradication [11][12][11,12]. These findings indicate that removal of the causative agent not only leads to interruption of fibrogenic signals, but also induces fibrolytic/restorative pathways, resulting in regression of fibrosis. However, a certain fraction of patients does not regress, suggesting a potential involvement of genetic/epigenetic mechanisms [13].
In experimental studies performed in mice treated with CCl4 to develop a pre-cirrhotic stage of liver injury and then allowed to spontaneously recover upon toxin withdrawal, resumption of CCl4 exposure rapidly induced profibrogenic features in HSCs, indicating that an “epigenetic memory” can be induced in these and, possibly, other cells [14][15][14,15].
3. Myofibroblast Clearance or Inactivation
The role of activated myofibroblasts in the development of liver fibrosis is well established. Different cell types can contribute to the myofibroblast population, including HSCs, portal fibroblasts, bone marrow-derived collagen producing cells (fibrocytes) and, possibly, parenchymal cells undergoing epithelial-mesenchymal transition (EMT) [16][49]. Although the origin of activated myofibroblasts may vary depending on the different etiologies of disease [17][50], HSCs can be considered their major source, as demonstrated by studies showing that HSCs depletion improves fibrosis in models based on both CCl4 intoxication and bile duct ligation [18][51]. Even in biliary fibrosis, where portal fibroblasts have been suggested to be the primary cell type initiating the fibrogenic response, giving rise to more than 70% of myofibroblasts, activation of HSCs becomes crucial after the initial phases [17][50]. During fibrosis regression, in response to a decrease of fibrogenic stimuli, the number of myofibroblasts drops, due to multiple mechanisms, that include restraint of activation, apoptosis, senescence, immune clearance, and reversal to a quiescent-like phenotype [14][15][19][20][21][14,15,52,53,54].4. Modulation of Inflammatory Processes
Inflammation represents a main feature of chronic liver diseases and plays a key role in any stages of the fibrogenic process, even during fibrosis regression. Inflammatory response involves multicellular interactions, dynamically regulated by a plethora of factors (e.g., soluble mediators, ECM components, pathogen-associated molecular patterns-PAMPs, damage-associated molecular patterns-DAMPs), acting in cell-specific fashion and aimed to restore liver architecture and function, but also leading to liver fibrosis when the noxious agent persists. Cell death is an early and primary inducer of chronic inflammation and fibrosis. Hepatocyte-derived apoptotic bodies stimulate the secretion of pro-inflammatory and profibrogenic cytokines from macrophages and promote activation of HSCs through induction of autophagy [22][23][24][100,101,102]. In addition, injured hepatocytes release DAMPs, such as ATP, phormyl peptides, High Mobility Group Box 1(HMGB1) [25][103] and cytokines such as IL-33 [26][104], which triggers HSC activation directly or indirectly, by promoting IL-13 release by innate lymphoid cells (ILC2). At the same time, inflammatory mediators secreted by infiltrating immune cells contribute to cell death, amplifying hepatic injury [26][104]. As major effectors of fibrosis, activated HSCs play a central role in inflammation, receiving a wide variety of stimuli from inflammatory cells and from hepatocytes, cholangiocytes and activated sinusoidal endothelial cells (SECs). Activated HSCs are highly responsive to inflammatory mediators which induce inflammatory pathways (such as NF-κB and AP-1) [27][28][105,106] and consequent secretion of cytokine/chemokines that act in autocrine and paracrine fashion. Inflammatory signals exert specific roles on HSCs, maintaining survival (IL-1β, TNFα, CXCL12) and the activated state (ILs and chemokines) [29][107], providing chemotactic stimuli for HSCs themselves or inflammatory cells (CCL2, CCL5, CXCL9, CXCL10, CX3CL1) and mediating the gut-liver axis crosstalk (toll like receptors (TLRs)) [27][105]. All these processes can contribute to positively or negatively modulate inflammatory responses and fibrogenesis, promoting fibrosis progression or regression. As modulators of liver fibrosis, immune cells exhibit a dual role, being able to contribute to both fibrosis progression and regression [30][31][108,109]. Danger signals generated in the site of injury lead to infiltration of circulating inflammatory cells (T lymphocytes, neutrophils, dendritic cells and monocytes) and activation of Kupffer cells (KCs) [30][31][108,109]. The release of a wide range of soluble mediators amplifies inflammation and stimulates the fibrogenic process. Upon removal of the cause of injury, the balance switches from pro- to anti-inflammatory/restorative pathways, promoting fibrosis resolution. This shift is achieved by rearrangements in the type of immune cell populations recruited, with a marked drop in intrahepatic T cells and blood-derived cells (NKT cells, monocytes) [32][110], and phenotypic modifications of certain cell types, mainly macrophages.5. ECM Degradation
Liver fibrosis is a dynamic process characterized by an unfavorable balance between ECM deposition and degradation. Degradation of ECM represents one of the most relevant aspects of fibrosis regression and requires activation of MMPs, macrophage phagocytic activity and downregulation of MMP-inhibitory molecules, such as tissue inhibitors of MMPs, TIMPs [7][10][7,10]. MMPs are the main matrix-degrading enzymes [33][62] and, according to substrate specificity, can be grouped in collagenases (MMP8, MMP1 and MMP13) which cleave native fibrillar collagens to gelatin, gelatinases (MMP2, MMP9), degrading a wide range of substrates including gelatin, collagens and, in some extent, elastin, metalloelastases (MMP12) and others (Table 1).Table 1.
Classification of human metalloproteinases (MMPs) and their function.
|
MMPs |
GROUP |
FUNCTION |
|---|---|---|
|
MMP1, MMP8, MMP13 |
Collagenases |
Cleavage of native fibrillar collagens to gelatin |
|
MMP2, MMP9 |
Gelatinases |
Degradation of a wide range of substrates, including gelatin, collagens and elastin |
|
MMP12 |
Metalloelastases |
Elastin degradation |