Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Francesco Borgia and Version 2 by Catherine Yang.
High-mobility Group Box 1 (HMGB1) is a nuclear protein that plays a key role in acute and chronic inflammation. HMGB1 is closely associated with cell survival and proliferation and may be directly involved in tumor cell metastasis development thanks to its ability to promote cell migration. HMGB1 is closely associated with cell survival and proliferation and may be directly involved in tumor cell metastasis development thanks to its ability to promote cell migration.
- melanoma
- HMGB1
- immunogenic cell death
- alarmins
- RAGE
- metastasis
1. Cutaneous Melanoma
1.1. Cutaneous Melanoma Clinical Types and Pathogenesis
Melanoma is considered one of the most aggressive forms of skin tumors, consisting of the abnormal growth of melanocytic cells. It has been increasing steadily as a global incidence over the past five decades [1]. Despite being a relatively rare skin cancer compared to the others (<5%), melanoma is the leading cause of skin cancer-related mortality [2]. Superficial spreading melanoma, lentigo maligna melanoma, nodular melanoma, and acral melanoma are the most common clinical types [3]. The histological classification of melanoma is useful in its diagnosis and is an important feature in defining cancer-related survival; however, its molecular subtypes are often determined by various somatic mutations [4]. Melanoma pathogenesis, defined as melanomagenesis, is based on the acquisition of sequential alterations in various genes and pathways controling metabolic or molecular mechanisms which regulate crucial cell function, survival, and replication rate [1]. When mutated, the genes involved in cancer can cause dysregulation of molecular processes with subsequent phenotypic manifestations. Main pathways leading to melanomagenesis include, but are not limited to, the mitogen-activated protein kinase (MAPK) pathway, which includes neuroblastoma RAS viral oncogene homolog (NRAS) and V-RAF murine sarcoma viral oncogene homolog B1 (BRAF), the cyclin-dependent kinase inhibitor 2A (CDKN2A) pathway, and the phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway. The recognition of these mutated genes led to the development of targeted therapies, which have mostly been represented by the RAS–RAF–MEK–ERK and PI3K–AKT signaling pathways [5].
1.2. Molecular Categories and Therapy
Skin melanoma is subdivided into three molecular categories based on gene expression: a proliferation oriented-one, driven by the SOX10–MITF pathway; an invasive one, which enhances the activity of epithelial-mesenchymal transition (EMT) and transforming growth factor-beta receptor 2 (TGFBR2); and an immune-mediated one which modifies the tumoral local environment. The three major oncogenes define three molecular forms of melanoma: BRAF-, NRAS- and KIT-mutants; a fourth subtype, also called the “triple wild-type” form, includes all the above [3]. From these data, it emerges that the molecular characterization of melanoma is necessary before the therapeutic choice, to obtain adequate clinical management and a patient-based therapy. Different molecular patterns require different types of treatment, so target therapies have been proposed over time. Target therapies are effective just with exon 15/codon 600 BRAF- mutant melanoma, adopting BRAF inhibitors in combination or not with MEK inhibitors. There is no approved drug for NRAS-mutant melanoma yet [6]. When facing triple wild-type melanoma, the only therapy option available is immunotherapy, represented by anti-programmed cell death protein 1 (PD-1) and/or anti-cytotoxic T cell antigen 4 (CTLA-4) antibodies [7]. Programmed death-ligand 1 (PD-L1) expression of tumoral cells and/or tumor-infiltrating immune cells is a pivotal requirement of anti- PD-1/PD-L1 treatment for several cancer types. However, there is no need in case of melanoma; in fact, clinical responses can be observed in a significant portion of PD-L1-negative cases [8]. Unfortunately, only adjuvant therapies are available for stages III and IV. They are represented by anti-PD-1 molecules, which are independent of mutational status, or dabrafenib and trametinib for BRAF mutant patients. The latest guidelines for the treatment of stage IV melanoma, either resected or not, state that systemic treatments are always mandatory. For first-line treatment, especially in BRAF wild-type patients, immunotherapy with PD-1 antibodies alone or in combination with CTLA-4 antibodies should be considered. In stage IV melanoma with a BRAF-V600 E/K mutation, first-line therapy with BRAF/MEK inhibitors can be proposed instead of immunotherapy [7].
2. HMGB1
2.1. Structure and Localization
High mobility group box 1 (HMGB1) is a nuclear protein, encoded by a gene on chromosome 13q12, that acts as a non-histone chromatin-binding protein, with 215 amino acids and a molecular weight of 25-kDa. HMGB1 is composed of two proximal homologous DNA binding domains and a C–terminal ending with residues of glutamic and aspartic acid [9]. HMGB1 is normally localized in the nucleus, but it can move to the cytosol, or it can be released or actively secreted in the extracellular space by injured cells. Inside the nucleus, HMGB1 binds chromatin, stabilizes DNA, and regulates gene transcription by binding/bending duplex DNA. After damage signals, it shuttles from the nucleus to the cytoplasm or is released, thus affecting multiple cell stress responses and inflammatory mechanisms [10]. The translocation of HMGB1 from the nucleus to the cytoplasm is driven by the nuclear localization signals (NLS1 and NLS2) and nuclear export signals (NESs). Exogenous stimuli, which cause injury or death, lead to cellular alarm system activation thus releasing alarmins or pathogen-associated molecular patterns (DAMPs) to signal danger-induced cellular stress [9][10][9,10].
2.2. Mechanism of Release and Receptors
Two HMGB1 release mechanisms are known: passive release and active secretion. HMGB1 is passively released from damaged or necrotic cells triggering an immediate inflammatory response via pro-inflammatory cytokines such as TNFα. Active secretion of HMGB1 occurs via immune cells, endothelial cells, platelets, neurons, astrocytes, and tumor cells during stress or secondary to other DAMP signals as reinforcement [11].
HMGB1 can bind to several extracellular receptors, such as the one for advanced glycation end products (RAGE), Toll-like receptor (TLR) 9, TLR4, TLR2, integrins, mucin domain (TIM-3), and C-X-C chemokine receptor type 4 (CXCR4) [9]. Given the plethora of possible target receptors for HMGB1, each domain can interact with different molecules and, in particular, residues 150–183 bind to RAGE, a major actor in the beginning and maintenance of the inflammatory process. RAGE, belonging to the immunoglobulin superfamily, is a transmembrane receptor with an extracellular domain, a transmembrane domain, and a cytoplasmic tail of 43 amino acids. While the extracellular domain is responsible for ligand binding, the cytoplasmic tail is involved in intracellular signal transduction [2]. Firstly, described as a receptor for advanced glycation end products (AGE), RAGE is now recognized as a multi-ligand receptor, including HMGB1. RAGE is involved in HMGB1-induced cell inflammation, proliferation, migration, and immunity. Furthermore, extracellular HMGB1 stimulates RAGE expression in several cytotypes [12].
2.3. Function
HMGB1 plays a key role in both acute and chronic inflammation. In physiological conditions, this protein is inside the nucleus of quiescent macrophages/monocytes. Cell stress or death are the main mechanisms that lead to the release of HMGB1 outside the cell membrane, thus HMGB1 functions as an alarmin, meaning a molecule that triggers an inflammatory response in combination with other cytokines, DAMPs, and pathogen-associated molecular patterns (PAMPs) [13]. HMGB1 plays a central role in autophagy induction, an endogenous survival mechanism against cell stress [14]. HMGB1 induces autophagy in the nucleus upregulating the expression of heat shock protein (HSP) 27, while in the extracellular space, once released by cancer cells, it binds RAGE inducing in turn autophagocytic activity in nearby cells [9]. It is demonstrated that cancer can upregulate autophagy, which leads to drug resistance thwarting chemotherapy [11]. HMGB1-induced autophagy can also be regulated by several miRNAs, a family of small non-coding RNAs which, through epigenetic mechanisms, play an important role in the maintenance of immune homeostasis, cancer progression, and inflammation. Recently, it was demonstrated that several miRNAs modulate HMGB1 expression and its functions [9]. Moreover, miRNAs can affect HMGB1 gene expression, modulating cancer progression. HMGB1 is a direct target of miR-548b, and the expression level of this miRNA can suppress melanoma cells’ growth by targeting the HMGB1 pathway [15].
3. HMGB1-Related Melanoma Growth
HMGB1, after being released from damaged or necrotic cells, is used by the immune system to recognize tissue damage in order to initiate repair responses and to promote lymphocyte maturation [16]. From here, thwe researchers ccan deduce a role in oncological diseases [16]. One of the important stages of disease progression is neoangiogenesis, which is fundamental for sustaining the metabolic and oxygen demands of cancer cells. It is known that solid tumors exhibit large hypoxic areas because of an imbalance between their oxygen supply and consumption. This hypoxic environment results in focal areas of tumor cell necrosis with consequent release of DAMPs and alarmins, including HMGB1 [17]. Extracellular HMGB1, released from the tumor cells under hypoxia, mediates communication between cells in the tumor microenvironment through binding several receptors, especially RAGE and TLR4, which contribute to tumor growth via sustenance of long-term inflammation [16]. Once secreted by melanoma cells, HMGB1 binds to RAGE, activating the endothelial cells with consequent increased expression of the adhesion molecules VCAM-1, ICAM-1, and E-selectin [16]. Within the tumor necrotic zones, the release of HMGB1 leads to the activation of kappa-light-chain-enhancer of activated B cells (NF-κB), which in turn upregulates leukocyte adhesion molecules and the production of pro-inflammatory cytokines and angiogenic factors, including vascular endothelial growth factor (VEGF) [16]. Tumor-Associated Macrophages (TAMs) are recruited in the tumoral milieu characterized by the presence of chemokines and proinflammatory factors [18][19][20][18,19,20]. After tumor infiltration, macrophages can acquire two different phenotypes: the M1 phenotype exerts a cytotoxic effect on tumor cells, with increasing production of nitric oxide (NO) and reactive oxygen species (ROS), which mediate the apoptosis of neoplastic cells [21]. Tumor cells create a microenvironment that promotes the acquisition of an M2 phenotype, which possesses pro-tumor characteristics, and, in contrast to M1 phenotype, exhibits low cytotoxic properties with defective production of NO and ROS, promoting the growth and vascularization of tumor cells [21][22][21,22]. HMGB1, released by melanoma cells, promotes the accumulation of M2-macrophages, which enrich the tumoral microenvironment with IL-10, turning down the tumoral killing [22]. Further evidence that the production of HMGB1/RAGE-dependent IL-10 by macrophages promotes melanoma growth has come from a human melanoma tissue study, in which high infiltration of IL-10-producing macrophages was detected in melanoma tissue with a high expression of HMGB1 [17]. Since HMGB1 directly induces IL-10 production in TAMs, blocking IL-10 with a neutralizing antibody led to delayed tumor growth in a B16 mouse melanoma model [17]. RAGE/HMGB1 axis also activates T lymphocytes, as demonstrated by a study with animal specimens: the study showed that the HMGB1/RAGE axis influences melanoma growth via the expression of IL-23 and IL-17 from a subpopulation of T cells, (γδ-T cells) [23]. Growth of melanoma cell line B16-F10 was significantly inhibited, and expression of IL-23 and IL-17 was markedly reduced in RAGE−/− mice compared with wild-type mice. The same study also showed that HMGB1 stimulates the production of IL-23 in a RAGE-dependent manner, which in turn promotes the expression of IL-17; subsequently, IL-17 promotes tumor growth through IL-6 induction with the consequent activation of the Signal transducer and activator of transcription 3 (STAT3) [23]. In addition to promoting carcinogenesis, interleukin production, and shift of T cell subpopulation, the HMGB1/RAGE axis has been shown to play a key role in suppressing cytotoxic T cell activity by increasing PD-L1 expression levels [24][25][24,25]. Moreover, HMGB1 levels were higher in patients who did not respond to the immune checkpoint inhibitor ipilimumab than in responding patients, supporting the hypothesis that the HMGB1/RAGE axis also leads to a tumor-promoting microenvironment [26]. In addition, RAGE was detected in the cytoplasm of human melanoma cells (G361 and A375) and the treatment with AGEs induced the proliferation and migration of human melanoma cells. Thus, treatment with anti-RAGE antibodies could explain the inhibition of tumor formation, invasion, and increase in the survival rate of specimens in an in vivo animal model [27]. AGEs and RAGE could be a valuable target in the near future for the treatment of melanoma since their levels are abundantly inferior in healthy skin, thus suggesting that this type of therapy could have potentially very few side effects [28]. The main pathways involved in melanoma growth are represented in Figure 1.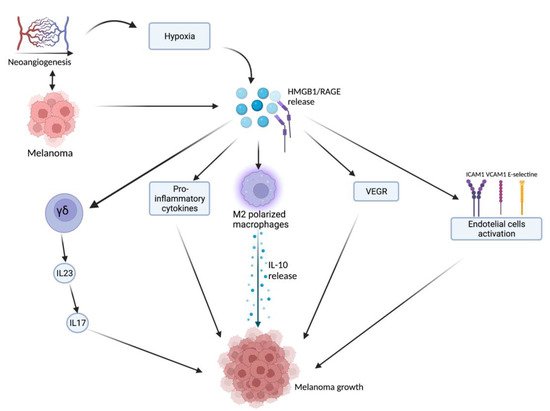
Figure 1. Tumor microenvironment and hypoxia, caused by cancer cells’ metabolic and oxygen demands, enhance HMGB1 release. HMGB1 binds to RAGE and actives several pathways: γδ-T cells, pro-inflammatory cytokines release, M2-macrophages, VEGF activation, and endothelial cell activation. All these lead to melanoma growth through the maintenance of an inflammatory microenvironment. Created with BioRender.com.