The phenotypic effects of single nucleotide polymorphisms (SNPs) in the development of sporadic solid cancers are still scarce. The aim of this review was to summarise and analyse published data on the associations between SNPs in mismatch repair genes and various cancers. The mismatch repair system plays a unique role in the control of the genetic integrity and it is often inactivated (germline and somatic mutations and hypermethylation) in cancer patients. Here, we focused on germline variants in mismatch repair genes and found the outcomes rather
controversial: some SNPs are sometimes ascribed as protective, while other studies reported their pathological effects. Regarding the complexity of cancer as one disease, we attempted to ascertain if particular polymorphisms exert the effect in the same direction in the development and treatment of different malignancies, although it is still not straightforward to conclude whether polymorphisms always play a clear positive role or a negative one. Most recent and robust genome-wide studies suggest that risk of cancer is modulated by variants in mismatch repair genes, for example in colorectal cancer. Our study shows that rs1800734 in MLH1 or rs2303428 in MSH2 may influence the development of different malignancies. The lack of functional studies on many DNA mismatch repair SNPs as well as their interactions are not explored yet. Notably, the concerted action of more variants in one individual may be protective or harmful. Further, complex interactions of DNA mismatch repair variations with both the environment and microenvironment in the cancer pathogenesis will deserve further attention.
- mismatch repair, genetic variants, genes, genotype
1. IntroductionDNA mismatch repair system and its role in tumorigenesis
1.1. DNA mismatch repair system and its role in tumorigenesis
DNA mismatch repair system (MMR) is an integral part of DNA damage response pathway (DDR), responsible for maintenance of genomic integrity. MMR preferably corrects frameshift mutations in microsatellites and mismatched nucleotides generated during DNA replication [1][2]. The importance of MMR in replication fidelity is illustrated in Table 1. Depending on the type of DNA damage, MMR also interacts with DDR and triggers therefore cell cycle arrest and apoptosis [3]. Impaired MMR function results consequently in DNA damage accumulation and mutational load, leading to DDR disruption and genomic instability. It has been postulated that every DDR process is functionally affected during carcinogenesis [4]. Defects in DDR genes are particularly apparent in hereditary and familial cancers with known high penetrance germline mutations in DNA repair genes [5][6][7][8][9]. For instance, loss of function of MMR (so called mutator phenotype) due to germline mutations is responsible for hereditary non-polyposis colorectal cancer (also Lynch) syndrome [6][10]. Genetic changes in MMR system underlie predisposition and etiopathogenesis to cancers of the colon, endometrium, ovary and other organs [8][9][11] , recently reviewed by Valle et al. [12]. While rather rare germline mutations in MMR (MSH2/MLH1) genes exert high effect in above hereditary syndromes, sporadic malignancies arise as an interplay of multiple common alleles (including those in MMR system) with rather modest effect [2][13]. Since the overwhelming majority of malignancies is of sporadic (non-familial) origin, these common variants in individual’s genetic material that predispose to higher cancer risk and its further progression are currently of importance. Variations in MMR genes are common in the general population and may be associated with a moderate increase in cancer risk [14]. Both coding and non-coding MMR genetic variants may disrupt the physiological function of MMR proteins; even alterations that do not cause a change in amino acid sequence could lead to aberrant splicing, changes in transcription regulation, modifying microRNA binding etc. [15]. All these variants are in important interplay with environmental, microenvironmental and lifestyle factors. Especially the combination of SNPs considered as less pathogenic in one individual may increase the risk of cancer in the others [16][17][18]. Finally, genetic variants may also modulate the response to therapy and drug toxicity [19].
Table 1. The DNA error frequencies during the replication. MMR appears important in ensuring the DNA replication fidelity [20].
|
Mechanism |
Error frequency |
|
Base pairing |
10-1 to 10-2 |
|
DNA polymerases (basection,seleproofreading, |
10-4 to 10-5 |
|
Accessory proteins |
10-7 |
|
Mismatch repair |
10-10 |
1.2. Function of DNA mismatch repair system
2. Function of DNA mismatch repair system
The S-phase of the cell cycle is characterized by DNA replication and the duplication of the whole genome. The replicative polymerases may incorporate inappropriate nucleotides leading to base-base mismatches or they can slip on nucleotide sequences, creating insertion-deletion loops (IDLs). This erroneous activity of DNA polymerases generates single nucleotide point mutations or frameshift mutations resulting in the synthesis of truncated protein. Arising biosynthetic errors in newly synthesized DNA, not corrected by exonucleases, are removed by MMR system. In procaryotes, it involves three specialized proteins: MutS, MutL and MutH. MutS is an ATPase acting as a homodimer or heterotetramer, which recognizes base-base mismatches and small IDLs [21][22][23]. Its loading onto the DNA leads to conformational change allowing its interaction with MutL - an ATP-dependent heterodimer, essential for removal of DNA fragment containing the mismatch [24]. Finally, MutL interacts with another MMR component MutH and activates its endonuclease activity. It nicks unmethylated strain of hemi-methylated DNA. MutH has no homolog in eukaryotes, its 5´→3´endonuclease activity is taken up by MutL homologs [25][26].
The eucaryotic MMR system (Figure 1) consists of following homologs: five MutS (MSH2-MSH6), two MutL (MLH1, MLH3) and three postmeiotic-segregated proteins (PMS1, PMS2, PMS2L). MSH2 forms a heterodimer with MSH6 or MSH3 giving rise to the assembly of MutSα and MutSβ complexes, respectively [27][28]. MutSβ complexes repair preferentially heteroduplexes with larger IDLs than MutSα complexes [29][30]. In humans, MLH1 is the major MutL homolog which assembles to MutLα, MutLβ and MutLγ complexes with PMS2, PMS1 and MLH3 proteins, respectively [31][32]. SNPs were identified practically in all MMR genes, predominantly in MLH1 and MSH2 (Figure 1).
In fact, the MMR system is so vital for the integrity of the human genome that low penetrance variations are strongly selected. It is very unlikely that these alleles can drift to polymorphic level. However, a combination of more polymorphisms with minute effect may disrupt cellular responses to DNA damage and alter individual´s sensitivity to carcinogens [16][33]. They may modulate tumour occurrence, growth, metastatic progression and response to chemotherapy. But there is still a gap in understanding the real function of polymorphisms in the human genome and their impact on the cancer development. Here we overview the possible role of SNPs in MMR genes in susceptibility and the development of cancer, as well as in the prognosis and treatment prediction.
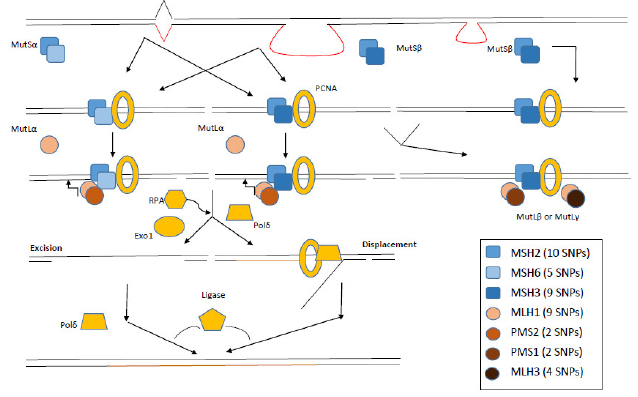
Figure 1. Molecular principles of eucaryotic MMR system. MutSα protein complexes recognize mainly nucleotide mismatches, whereas MutSβ insertion-deletion loops. Further steps of MMR include interaction with MutL protein complexes and other proteins (exonucleases, replication proteins and polymerases). Numbers in brackets summarize the number of known pathogenic SNPs with F ≥ 5 % in EUR population (adapted from [34][35]). PCNA represents Proliferating cell nuclear antigen, EXO1 Exonuclease 1, Pol δ DNA polymerase δ and RPA Replication protein A.
23. Materials and methods
PubMed database was searched for the results of all studies published before July 2020, using the keywords “mismatch repair gene”, “polymorphisms”, “genetic variant”, “sporadic cancer”, “single nucleotide polymorphism”, “genome-wide association studies”. Relevant papers were selected and critically analysed. The dbSNP database (http://www.ncbi.nlm.nih.gov/snp/) was used as the primary source of all SNPs. All human MMR genes were also analysed for SNPs in open-access SNP databases, when additional information was needed: F-SNP database of Queen´s University (http://compbio.cs.queensu.ca/F-SNP/), Canadian MutDB of The Buck Institute Mooney Lab (http://mutdb.org/) and in British In-SIGHT (The International Society for Gastrointestinal Hereditary Tumours Incorporated) database (http://www.insight-group.org/). Relevant papers cited in these databases were also assessed and used in the review. We have concentrated on papers describing the role of SNPs in human MMR genes in the development of primary sporadic solid cancers. We prepared our own database that included those selected SNPs with their frequency in all populations worldwide. In this review, however, we focused on European population primarily and considered the minimal population frequency of SNP exceeding 5 %. We also considered the pathogenicity of SNPs, which were studied in non-European populations only, but their frequency in the European population is equal to or more than 5 %. All other SNPs, even pathogenic in other populations, but with lower allele frequency in the European population, were not included in this review. Additional information about SNP reference numbers, gene location, amino acid change, phenotypic change prediction, minor allele frequency in the European and other populations and references to literature, is available in the Supplement. All SNPs, either with an adverse or protective effect on the onset of cancer, were summarised by Venn´s diagrams. SNPs that had the same impact for various types of cancer were localised in the centre of the diagram, SNPs with more specific impact were localised more peripherally. This review focuses primarily on genes encoding parts of MMR protein complexes MutL and MutS, other MMR-related proteins, such as Proliferating cell nuclear antigen (PCNA), Exonuclease 1 (EXO1), DNA polymerase δ (Pol δ), Replication protein A (RPA) have not been considered. The workflow of this study is presented in Figure 2. The functionality of SNPs was ascertained by using in silico approaches. The phenotypic effect of the SNP on the functionality of the protein was inferred on the basis of software, such as SIFT, PolyPhen, PupaSuite.
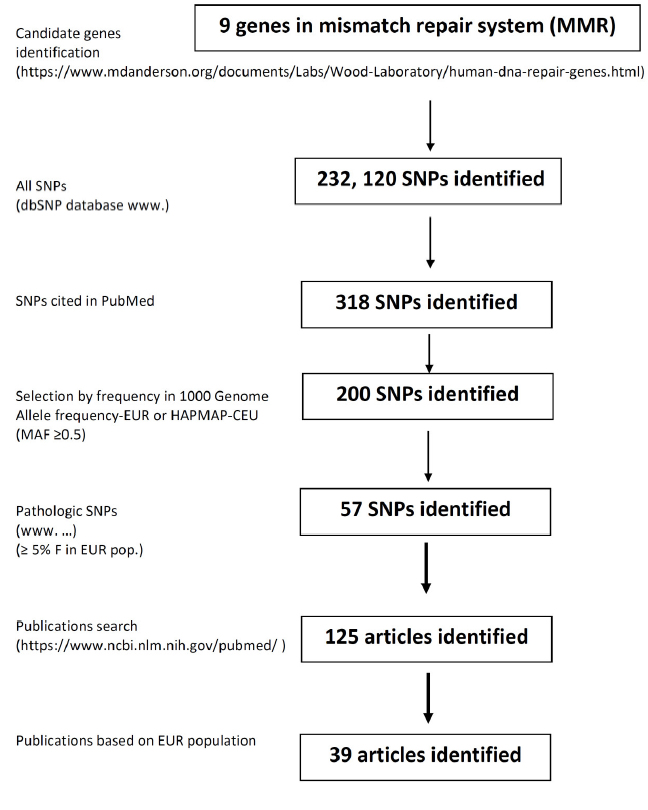
Figure 2. Workflow of the study.
34. Results on MMRLH1 gene varian(muts
3.1.L MLH1 homologene (mutL homolog 11)
MLH1 gene, located on chromosome 3 at the p22.2 site, encodes substantial protein of the MMR system. Twenty-three transcripts have been identified so far. The longest NM_000249 is coding for a 756 amino-acid protein MLH1, which forms heterodimers with other MMR proteins, including PMS2, PMS1, and MLH3. MLH1 is the most frequently mutated MMR gene, in both sporadic and hereditary cancers. MLH1 gene mutations represent the most prevalent cause of an inherited form of colorectal cancer (CRC), hereditary non-polyposis colorectal cancer HNPCC [12][36]. According to the comprehensive dsSNP server human MLH1 gene contains 15, 721 SNPs. But only 49 SNPs were cited in PubMed database.
Apart from coding SNPs and variants in regulatory regions for corresponding gene expression, there are MMR variants that may also interact with epigenetic silencing (e.g. methylation), contributing further to gene expression regulation with implications on the MMR system [37].
SNPs located in the promoter region of the MLH1 gene are sufficient to affect the expression of the gene. For example, the most intensively studied intron SNP rs1800734 (c.-93G>A, upstream variant near 5´ untranslated region) was repeatedly described as pathogenic, increasing the risk of various types of cancer, sporadic CRC and endometrial cancer in particular [38][39][40][41][42][43]. The SNP is located in the region essential for the maximum transcriptional activity. For instance, the A/A genotype was associated with the increased risk of glioblastoma (odds ratio (OR)=3.14, 95% confidence interval (CI)=1.09-9.06, P=0.034) [44]. This SNP also increases the risk of lung adenocarcinoma in European, Korean and Taiwanese populations [45][46][47][48]. One European study described the association between the A/A genotype of this SNP and lung cancer in interaction with chromium exposure [48]. The accumulated evidence on the pathogenicity of this SNP in the lung cancer is highlighted in a meta-analysis, comprising 1, 018 publications and 754 genes [49]. The precise mechanism by which rs1800734 down-regulates the function of MLH1 is unknown, but some mechanisms have been suggested. The variant is localized in the promoter region in the consensus sequence binding transcription factor AP-4, important for the initiation of transcription [47]. Raptis et al. found this variant in two patients with microsatellite instability-high (MSI-H) form of CRC. The rs1800734 variant was also associated with increased promoter methylation suggesting the existence of the sequence specificity for promoter methylation [50]. Indeed, the risk allele of the SNP rs1800734 showed a strong association with MLH1 promoter hypermethylation and loss of MLH1 protein in CRC tumours [37]. Recently, Ning Qing Liu et al. reported the A-allele of rs1800734 within the promoter region of MLH1 as perturbing the binding of allele-specific transcription factor AP4 and consequently increasing DCLK3 expression through a long-range interaction, which promotes malignant transformation through enhancing expression of the genes related to epithelial-to-mesenchymal transition [51]. An additional study confirmed that rs1800734 is a functional polymorphism that results in decreased MLH1 transcription [52]. However, it has to be considered that different effects of SNPs on the function of the gene may emerge in different human populations. On the other hand, Pan et al. proved in a meta-analysis comprising the data from 17 studies that SNP rs1800734 was not associated with an increased risk of cancer [53]. The authors noted following peculiarity: low-quality studies were associated with a higher risk than high-quality studies and hospital-based studies showed a higher risk than population-based studies. Interestingly, a more pronounced risk was observed in the Asian population than in the Caucasian population. The divergent results were provided by the studies on CRC patients. While rs1800734 increased the risk of CRC, the same SNP decreased the risk of rectal cancer alone [54]. The rs1800734 polymorphism within the core promoter region of MLH1 gene has been reported to increase a risk of microsatellite instable (MSI-H) CRC (OR = 1.39; P = 1.45 × 10-4), as assayed for in 3, 132 cases. A meta-analysis strengthened further the above evidence for rs1800734 as an event in a specific CRC subtype (P = 3.43x10-12) [40]. Finally, recent study based on genome-wide association studies (GWAS) data by Pardini et al. comprising over 27, 000 individuals showed rs1800734 to be significantly associated with the risk of colon cancer (OR=1.13, 95% CI=1.07-1.18, P=3.48x10-6) [55]. When looking at tumour site, rs1800734 was mainly associated with proximal colon tumours (OR=1.13, 95% CI=1.07-1.18). Due to many differences in embryogenesis, etiology, anatomy, genetics, and treatment response between the colon and rectal tumours, CRC is no longer a single etiopathogenic entity. The studies reporting the association of rs1800734 with cancer are provided in the Supplement. The summary of adverse effects of SNPs in MLH1 is shown in Figure 3. Our recent review pointed out that promoter hMLH1 hypermethylation is associated with CIMP (CpG island methylation phenotype) and acts as main mechanism for the evolution of sporadic MSI CRC [56]. Interestingly, the rs1800734 polymorphism within the core promoter region of MLH1 gene has been reported to increase a risk of microsatellite instable (MSI-H) CRC, accounting for about 20% of cases. Variation in 5´-UTR region of MLH1 (Table 2) is likely to be associated with methylation status of this gene (variant allele linked with hypermethylation) and increase the risk of various malignancies (Figure 3). Whether this effect is mediated sterically by variation or via the altered activity of DNA methyltransferase remains to be elucidated.
Additionally, analysis of a region of chromosome 3p21 spanning the MLH1 locus in peripheral blood cells of healthy individuals showed that a CpG island shore 1 kb upstream of the MLH1 gene evinces differential methylation profile following the stratification by MLH1 variants (rs1800734, rs749072, and rs13098279). Significantly higher white blood cell shore methylation has been reported in individuals with wild-type genotypes as compared to those with heterozygous or homozygous genotypes [57]. Although the differences in MLH1 promoter methylation may not solely explain the increased risk of several cancers exerted by rs1800734, this epigenetic mechanism should not be overlooked.
Another intensively studied SNP in MLH1 gene, rs1799977 (c.655A>G; Ile219Met), is located in exon 8 [58]. This SNP was often ascribed as harmful and associated with increased risk of various types of cancer (summarised in the Supplement). Nejda et al. found that rs1799977 elevates the risk of sporadic CRC but it also provides better outcomes for CRC patients: G allele carriers displayed a lack of vascular invasion, less frequent distant metastases, and reduced recurrence than wild-type A carriers [59]. Taken together, rs1799977 is considered as a SNP associated with the increased risk of CRC, breast (BC) and lung cancer, invasive, but not serous, ovarian cancer, prostate cancer, and also with the increased death risk from B-cell lymphoma [41][60][61][62][63][64]. On the contrary, some authors found that homozygous carriers of rs1799977 had also lower risk of BC and lung cancer in a Caucasian population [60][65]. Although SNP rs1799977 has no significant impact on the MLH1 function in vitro, it is associated with many functional defects in vivo. For instance, Campbell et al. proposed that it is statistically significantly associated with the Western diet and smoking (P=0.03 and P=0.005, respectively) [66]. Patients with Ile/Ile amino acid change, who consumed more red and processed meat, eggs, high-fat foods, refined grains and added sugars, conferred a higher risk of CRC. In summary, it is evident that SNP rs1799977 should be considered individually in each population for its impact on cancer risk. For example, the investigation of rs1799977 in almost 1, 000 patients of the Czech origin, which became a part of extensive international meta-analysis, showed no association with the risk of sporadic CRC [67]. However, this SNP should be analysed separately in patients with colon and rectal cancer as discussed earlier. It was observed that rs1799977 carriers have a higher risk of CRC and display the expression imbalance of MLH1, but the same SNP has a protective effect in rectal cancer patients [41][59][68].
Interesting results were obtained from the multi-gene studies. The role of SNPs in various genes associated with the CRC risk was intensively studied in the Czech population due to the high incidence of the disease in this area. The study of Tulupova et al. included five MMR genes which were genotyped in 614 cases and 614 matched controls for 10 SNPs. For MLH1, the authors focused on an intronic variant rs4647269 (c.791-1406C>T) and observed that its carriers exhibited a decreased risk of rectal cancer (OR=0.71, 95% CI=0.51–0.9, P=0.04) [54]. The same study found an intronic SNP of MSH6 rs3136228 (c.-152-405T>G) to be associated with increased risk of CRC (OR=1.29, 95% CI=1.02–1.62) and exonic rs1042821 (c.116G>C, Gly39Ala) with decreased risk of CRC (OR=0.76, 95% CI=0.60–0.98). Rs2072447 (c.3438+14A>T) was associated with increased risk of colon cancer (OR=1.34, 95% CI=1.03–1.74). Authors observed a differential distribution of haplotypes based on three MSH6 SNPs in the cases and controls (global P=0.02). The TAG haplotype was associated with a decreased risk of CRC (OR=0.74, 95% CI=0.59–0.92), whereas the most frequent haplotype GGG with increased risk of rectal cancer (OR=1.32, 95% CI=1.05–1.65) [54]. Another population-based study analysed the association of SNPs in MMR genes with prostate cancer risk [62]. A population (1, 484 cases) included Caucasian and African-American men, who were diagnosed with prostate cancer between 1993 and 2005. Among Caucasians, the authors disclosed a strong association between rs9852810 (c.1410-1306G>A) and overall prostate cancer risk (OR=1.21, 95% CI=1.02-1.44, P=0.03), more aggressive prostate cancer (OR=1.49, 95% CI=1.15-1.91, P<0.01) and prostate cancer recurrence (hazard ratio (HR)=1.83, 95% CI=1.18—2.86, P<0.01). The authors also reported more aggressive prostate cancer in association with rs1799977 [62]. This SNP was not analysed for its pathogenicity in prostate cancer in other population so far. A marginal association between a risk of squamous cell type lung cancer and intronic variants rs4647250 (c.453+544T>C), rs1558528 (c.67+955C>A) and rs2286939 (c.1038+86T>C) was observed in Chinese population, P=0.04 for all these SNPs [61]. The same study also found the association between the G allele of rs1799977 and the increased risk of lung cancer (OR=3.65, 95% CI=1.44-9.24, P=0.006); particularly among younger patients (OR=5.28, 95% CI=1.45-19.21, P=0.01). Only few SNPs had the protective effect in various cancers, for example rs2286940 (c.1410-169C>T) was associated with the better overall survival (OS) of lymphoid cancer patients (HR=2.06, 95% CI=1.35-3.16, P<0.001) and rs1540354 (c.307-1403A>T) with the better OS of CRC patients (HR=1.90, 95% CI=1.14-3.17, P=0.01) [69][70]. A GWAS study on 2, 795 predominantly Caucasian women and 4, 505 controls addressed SNP-SNP interactions (based on a priori knowledge for potential epistatic interactions) in BC susceptibility. The authors observed two-way SNP-SNP interactions (APEX1-rs1130409 and RPAP1-rs2297381; MLH1-rs1799977 and MDM2-rs769412) that conferred elevated risks for BC (P(interaction)<7.3 × 10-3) [71]. Figure 3 illustrates that SNPs rs1799977 and rs1800734 in MLH1 exert adverse effects on the risk of several cancer, except for squamous cell carcinoma (SCC). No consistent tendency may be tracked regarding the protective effects of MLH1 polymorphisms.
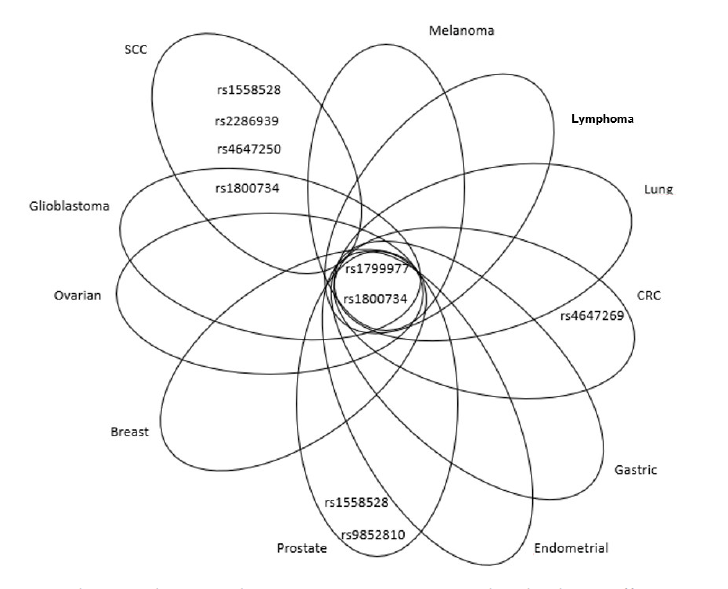
Figure 3. Venn diagram showing relevant MLH1 SNPs, associated with adverse effects, in overlap among different cancer types or peculiar for specific malignancies.
SCC=squamous cell carcinoma,
CRC=colorectal cancer
MLH3 gene (mutL homolog 3)
MLH3 gene is located on chromosome 14 at q24.3 site. Thirteen transcripts of this gene have been described so far. The longest one is coding a 1, 453 amino-acid protein MLH3, which forms a heterodimer with MLH1 (MutLγ complex), involved in meiotic crossing-over [72]. The other two MutL complexes, MutLα and MutLβ, do not consist of MLH3. According to the dbSNP server, 9, 007 SNPs were identified in human MLH3, but only 15 SNPs were cited in PubMed database. The majority of MLH3 gene SNPs is localized in extraordinarily long exon 2, which contains 3, 343 base pairs (bps). SNP rs175080 (c.2531C>T, Leu844Pro) was studied more exclusively. Conde et al. found that either homozygous or heterozygous variant was associated with decreased risk of BC [73]. Authors also recorded that combination of rs175080 with a SNP in MSH4 gene rs5745325 (c.289G>A, Ala97Thr) is pathogenic, being associated with an increased BC risk. MSH4 is a meiosis-specific protein and probably MLH3-MSH4 interaction is crucial for meiotic recombination [74]. The abrogated interaction between these two MMR proteins may influence the recombination process, resulting in an increased recombination rate in mammary gland cells. Interestingly, another study described rs175080 SNP as variant increasing the risk of lung cancer, but in interaction with other risk factors, such as smoking [75]. The association between AA genotype of rs175080 and a higher risk of the primary hepatocellular carcinoma was observed in the Chinese population [76]. This SNP increased the risk of cervical carcinoma, as assayed for on Chinese population as well [77]. Comprehensive pathway-based analysis of the combined dataset of two pancreatic cancer GWAS, PanScan 1 and PanScan 2, found an intronic SNP rs175057 (c.4012-37G>A) to be associated with the increased risk (P=0.039) of pancreatic cancer in the US population [78]. A GWAS performed on the Chinese population found an association of rs28756990 (c.2221G>A, Val741Ile) with the prostate cancer risk [79]. All other known SNPs in MLH3 were not studied for their role in the carcinogenesis. The overview of adverse effects of MLH3 SNPs for individual malignancies is shown in Figure 4: rs175080 has an adverse effect on hepatocellular, lung and cervical cancers, whereas rs28756990 and rs175057 were associated with prostate cancer only. Interestingly, rs175080 had a protective role in BC.
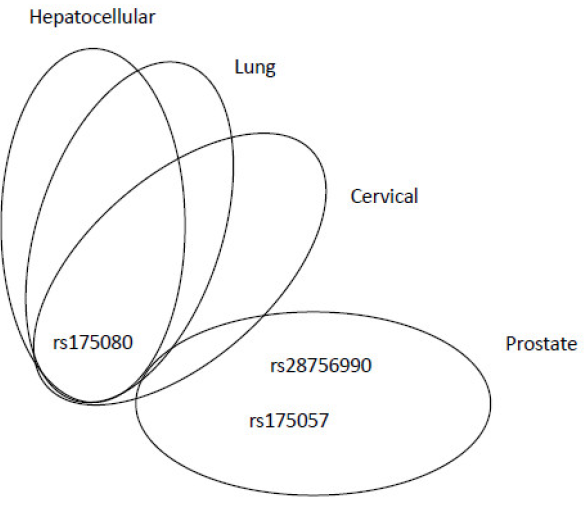
Figure 4. Venn diagram showing relevant MLH3 SNPs, associated with adverse effects, in overlap among different cancer types or peculiar for specific malignancies.
MSH2 gene (MutS homolog 2)
MSH2 gene is located on chromosome 2, at p21-p16.3 site. There are 6 transcripts of the gene, the longest one with 16 exons encodes a protein MSH2 encompassing of 934 amino acids. The dsSNP database comprises 68, 322 SNPs in MSH2, 83 of them cited in PubMed. Functionally, MSH2 is one of the most important protein components of post-replicative DNA mismatch repair system. Together with MSH6 and MSH3 it forms MutSα and MutSβ complexes, respectively. Briefly: MutSα repairs single-base mismatches or dinucleotide insertion-deletion loops, whereas MutSβ restores of long insertion-deletion loops. MutS complexes associate with MutLα heterodimers, involved in downstream repair events. MutSα also participates in the DNA homologous recombination repair and it regulates in melanocytes cell cycle regulation and apoptosis [80][81].
No exonic SNP in MSH2 was associated with the cancer risk (see Supplement). A recent study revealed the association between SNP rs2303425 (c.-68-50T>C), localised in 5´ UTR region, and increased risk of lung cancer (OR=2.28, 95% CI=1.12-4.63, P=0.024) in the Slovak population [82]. The same study showed on 422 cases and 486 controls that rs1800734 in MLH1 was associated with increased lung cancer risk (OR=1.40, 95% CI=1.08-1.82, P=0.01), the interaction between rs1800734 and rs2303425 (MSH2) revealed elevated risk for genotype GG/CC (OR=3.08, 95% CI=1.09-8.72, P=0.03), which was further pronounced in females (OR=11.56, 95% CI=1.33-100.36, P=0.005). The SNP rs2303425 in MSH2 is also associated with the higher risk of the luminal A subtype of BC in a Taiwanese population [83]. Individuals bearing C/C genotype had a significantly higher risk of BC compared to those with the T/T genotype (OR=2.0; 95% CI=1.1-3.8, P<0.05). The functional effect of this SNP on MSH2 expression was studied by luciferase assay, which showed that the cells with C/C genotype had significantly reduced expression of the MSH2 gene. The authors discussed that this SNP is located in NF-Y transcription factor binding site, also known as an estrogen-responsive element and might reduce the binding of the estrogen to the promoter of MSH2, resulting in its decreased activity [83].
Tumours of the gastrointestinal tract (GIT) are associated with the SNP rs1981929 (c.1277-118G>A), where variant allele confers increased risk of CRC, rs2303426 (c.211+9C>G) was associated with the gallbladder, oral and head and neck cancers, rs10183143 (c.1661+90T>C) posed a risk for gastric cancer [84][85][86][87][88]. The gastric cancer risk was further increased by drinking alcohol and eating the pickled and fried food [88].
An intronic SNP rs2059520 (c.2006-265A>G) was found to be associated with endometrial cancer risk (P=0.03) [39]. Increased risk of this type of cancer was also observed for SNP rs2303428 (c.2006-6T>A). The authors showed that women with binary SNP interaction of rs2059520 or rs2303428 with rs1800734 in MLH1 (accounting for about 9% of all endometrial cancer cases) had a particularly higher risk of this cancer, compared to those bearing wild-type (WT) variants of these SNPs (OR=2.1, 95% CI=1.2-3.6, P=0.005) [39]. However, both associations (rs2059520 or rs2303428) were confined to heterozygous genotypes; neither of the homozygous variant alleles was associated with endometrial cancer and this fact precludes clear interpretation. Rs2303428 is the most studied variant in MSH2 gene, being associated with various types of cancer (CRC, melanoma, lung, non-Hodgkin lymphoma, glioblastoma, and BC; for details see the Supplement).
There are no exonic SNPs in MLH2 that are associated with the risk of solid cancers. However, also SNPs localized in the intronic sequences, may significantly impact the development of cancer, OS and the efficacy of the treatment. The studies on MLH2 gene demonstrated how important role the genetic variants in the non-exonic sequences of the genome might play in human malignancies. The overview of adverse and protective effects of SNPs is shown in Figure 5 and Figure 6, respectively. The most intensively studied rs2303428 was found to be associated with seven different types of cancer. The other SNPs were considered as risk factors in fewer cancer types (Figure 5). Again, rs2303428 had protective effects in more types of cancer, suggesting that one SNP may have both adverse (Figure 5) and protective role in cancer (Figure 6). The reason of this ambiguity is still unknown, possible explanations are complex gene-gene and gene-environment interactions.
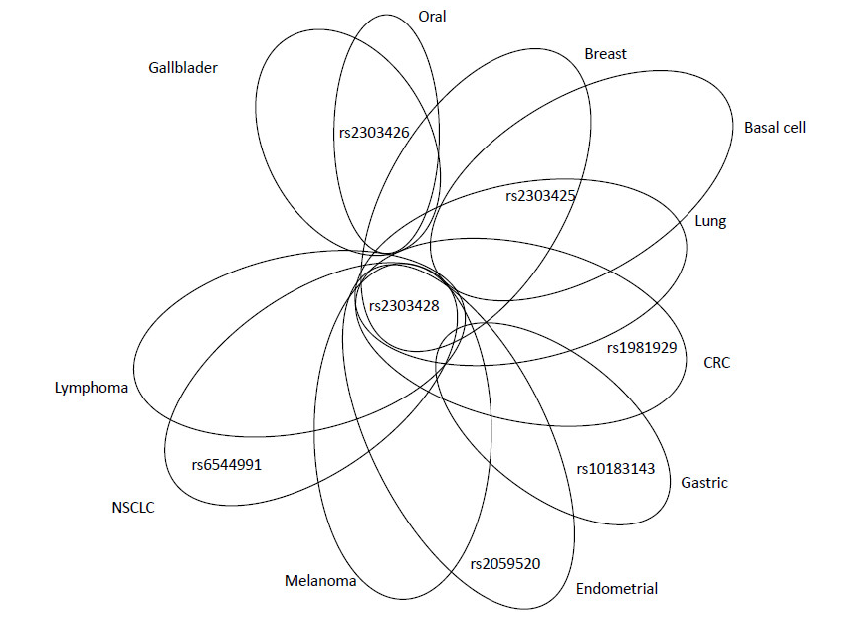
Figure 5. Venn diagram showing relevant MSH2 SNPs, associated with adverse effects, in overlap among different cancer types or peculiar for specific malignancies.
CRC=colorectal cancer,
NSCLC=non-small-cell lung cancer
Figure 6. SNPs of MSH2 with protective effects for the development of various tumours.
CRC=colorectal cancer,
NSCLC=non-small-cell lung cancer
MSH3 gene (MutS homolog 3)
MSH3 gene is located on chromosome 5 at q14.1 site. There are three known transcripts of MSH3, the longest one, consisting of 24 exons, encodes 1, 137 amino acid-long protein. The dsSNP database summarizes 53, 628 SNPs in humans, 46 of them were cited in PubMed. MSH3 forms a heterodimer with MSH2 to generate a MutSβ protein complex. MutSβ is involved in post-replicative DNA MMR. It initiates binding to mismatch and then forms an interaction with a MutLα protein complex. In general, MutSβ preferentially recognizes large insertion and deletion loops, up to 13 nucleotides long. The somatic mutations can be found in approximately 50 % of MMR-deficient CRCs [89]. MSH3 also interacts with the proliferating cell nuclear antigen (PCNA), which is important for the localization of MutSβ into replication foci. PCNA is assisting in the initiation step of MMR, guiding the MutSβ complex and other components of the MMR system to free termini in newly replicated DNA [90]. Interestingly, over-expression of MSH3, which can also occur in the tumour, causes substantial changes in the levels of MutSα and MutSβ complexes. MutSα is depleted and MSH6 is degraded. This can be explained by high levels of MSH3, which sequester MSH2, important for the formation of MutSα complex [91].
A non-synonymous SNP rs184967 (c.2846A>G; Gln940Arg) was found to be associated with the risk of proximal colon cancer (P=0.005). Individuals bearing this SNP along with higher intake of processed meat did not exert higher risk of CRC than individuals with WT variant. Patients with either the heterozygous G/A (OR=1.10, 95% CI=0.82-1.47) or homozygous A/A genotypes (OR=1.26, 95% CI=0.96-1.64) had worse progression-free survival (PFS) than those with G/G genotype, (OR=1.0; 95% CI=15.03-37.71), (P=0.04) [92]. One of the most studied SNPs in MSH3 is a frame-shift-coding SNP rs26279 (c.3133G>A; Ala1045Thr). Genotype frequencies of this variant were found to be higher in CRC patients (P=0.04) [93]. In the Portuguese population, gene-gene interaction between MSH3 Ala1045Thr and MSH6 Gly39Glu conferred the decreased BC risk (P=0.01) [73]. Several studies by analysing rs26279 observed increased risk of CRC, squamous cell cancer (SCC), non-small cell lung cancer (NSCLC), BC, hepatocellular carcinoma, and oesophageal cancer (see Supplement). In the meta-analysis authors assessed 11 publications comprising 3, 282 cases and 6, 476 controls and confirmed that this SNP increases the risk of various types of cancer, mainly CRC and BC, especially in Europeans and Asians [94], Supplement. An intronic SNP rs863221 (c.1763+1841T>G) was studied as prognostic marker on the cohort of 2, 060 CRC patients from the European population using SNP tagging approach. Among 68 SNPs in 7 MMR genes, 10 appeared significant in the univariate analysis (P<0.05), but after adjustment for sex, age, stage and diagnosis, only rs863221 in MSH3 gene remained associated with better OS in CRC patients (HR=0.59, 95% CI=0.42-0.82, P=0.001) [95]. Two additional SNPs (rs836808, rs1105524) were associated with an increased risk of BC and NSCLC. The first study discovered that an intronic SNP rs836808 (c.1174-176G>A) was associated with a higher risk of BC patients, if in interaction (1.06–1.19, P=0.008). This multi-centric consortium project collected data from 28 publications, including 70, 917 SNPs, 46, 450 patients and 42, 461 controls of European origin [96]. The second SNP (rs1105524, c.-35A>G) was studied on Chinese NSCLC patients, treated with platinum-based chemotherapy. Patients with either the heterozygous G/A or homozygous A/A genotypes had worse PFS (median survival time 14.3 months, 95% CI=9.80-18.75) than patients with G/G genotype, (median survival time 26.4 months, 95% CI=15.03-37.71), (P=0.04) [97].
A significant association between rs33003, an intronic SNP (c.3303-436A>G), and diffuse large B-cell lymphoma was found in a case-control study of subjects of European ancestry (P=0.01) [98]. This study encompassed 118 genes, 599 SNPs and 1, 116 samples. However, the authors discussed that this association was not replicated by another North American study [99], indicating this result as false positive. The overview of adverse effects of MSH3 SNPs is presented in Figure 7: the most studied rs26279 was associated with worse outcome of various cancer types. However, rs26279 had the protective effect on the development of BC. It seems that the evolution of BC differs in some molecular mechanisms from other types of cancer, with the significant protective role of SNPs in MMR genes. Rs836808 had opposite, adverse effect on the BC development.
Figure 7. Venn diagram showing relevant MSH3 SNPs, associated with adverse effects, in overlap among different cancer types or peculiar for specific malignancies.
CRC=colorectal cancer,
SCC=squamous cell carcinoma, NSCLC=non-small-cell lung cancer
MSH4 gene (MutS homolog 4)
MSH4 gene is located on chromosome 1 at p31.1 site. There is only one transcript of the gene identified, consisting of 20 exons and encoding a 936 amino acid-long protein. The dsSNP database covers 28, 103 SNPs in humans, but only 10 of them were cited in PubMed. The physiological functions of MSH4 protein are similar to those of MSH5: MSH4 is important for meiotic recombination, specifically required for reciprocal recombination and segregation of homologous chromosomes during meiosis I [100]. In the yeast Saccharomyces cerevisiae Msh4 and Msh5 act specifically to facilitate crossovers between homologous chromosomes [101]. The Msh4/Msh5 complex binds and stabilizes double Holliday junctions and promotes their resolution into crossover products. The ortholog of MSH4 in Caenorhabditis elegans, him-14, is important for crossing over in meiosis and loss of him-14 results in lack of chiasmata between homologs and consequent mis-segregation [102]. MSH4 forms a unique and exclusive heterodimer with MSH5, but also with MLH1 and MLH3 [74][103][104][74, 103, 104]. It appears that MSH4 acts during meiosis to direct the recombinational repair of DNA double-strand breaks produced in prophase I of meiosis I [105] and MSH4-MSH5 dimers important for mitotic and meiotic DNA double-strand break (DSB) repair [106][107]. MSH4 participates in the maintenance of genomic stability by restricting the use of error-prone non-homologous end joining following DSBs [108].
Among all SNPs cited in PubMed, only rs5745325 (c.289G>A; Ala97Thr) was associated with the increased risk of BC [73]. Interestingly, this SNP alone has no impact on the BC risk, but the authors described a gene-gene interaction between MSH4 Ala97Thr and MLH3 Leu844Pro (rs5745325 and rs175080 respectively) resulting in an increased BC susceptibility (A/G genotype of MSH4 with A/A genotype of MLH3 [OR=2.35, 95% CI=1.23-4.49, P=0.01], G/G genotype of MSH4 with A/A genotype of MLH3 [OR=2.11, 95% CI=1.12-3.98, P=0.02], and G/G genotype of MSH4 with A/G genotype of MLH3 [adjusted OR=1.88, 95% CI=1.12-3.15, P=0.02]). The study was conducted in a Caucasian Portuguese population.
MSH5 gene (MutS homolog 5)
MSH5 gene is located on chromosome 6 at p21.33 site. There are 19 transcripts of the gene, the longest one contains 25 exons, encoding an 834 amino acid-long protein. The dsSNP database comprises 6, 385 SNPs in humans, 22 of them were cited in PubMed. All findings in mice and humans indicate that MSH5 plays an important role in the meiotic recombination. Mice with a null MSH5 mutation (MSH5-/-) are viable but sterile. Phenotypically they display a disruption of chromosome pairing leading to defects in prophase I of meiosis [109]. The experiments in Caenorhabditis elegans showed that mutations in MSH5 did not affect the repair capacity of the MMR system, but they caused defective crossovers between homologous chromosomes [110]. However, recent studies showed that MSH5 is also important for the stability of mitochondrial genome after oxidative stress and for the repair of double-strand break (DSB) after chemotherapeutic treatment [111][112]. Since MSH5 interacts with other MMR proteins, it is a part of the MMR system [103].
There are only a few SNPs in MSH5 with significant implications in the neoplasia. The G allele of rs707938 (c.2148A>G; Gln716Gln) was proved in one study to be associated with the increased risk of lung cancer [113]. The authors also described patients with either homozygous variant A/A or heterozygous A/G variant of rs3131379 (c.813-45G>A) as susceptible towards lung cancer, this tendency was more pronounced among individuals who carried UNG (uracil DNA glycosylase) rs246079 SNP (c.802-574A>G). Other four original studies confirmed that rs3131379 is strongly associated with the lung cancer (see Supplement). Above finding was summarized in the meta-analysis by Timofeeva et al., which included 14, 900 cases and 29, 485 controls of European descent, based on 16 previously reported GWAS studies undertaken by nine analytical centers [114]. Yi et al. found by the yeast two-hybrid assay that the presence of SNP rs2075789 (c.85C>T; Pro29Ser) significantly weakens the interaction of MSH4-MSH5 complex [115]. The last two studies carried on Chinese population revealed that rs707938 increased the risk of hepatocellular carcinoma; rs707939 (c.1326+36C>A) had the negative effect in lung cancer patients, increasing the overall toxicity to platinum-based chemotherapy [76][116]. Recently, the authors investigated the role of DNA repair genes in LC patients by a multilevel association study with 1655 single nucleotide polymorphisms (SNPs) in 211 DNA repair genes on 6, 911 individuals pooled from four genome-wide case–control studies. Single SNP association corroborates association described in previous reports for MLH5 rs3131379 (P=3.57x10-5) [117]. Additional evidence on the concerted function of MSH5 gene variant with a decreased CpG methylation was reported for LC patients [118].
A GWAS was conducted in 1,952 LC cases and 1,438 controls. By pooling data with two other GWAS (5,095 cases, 5,200 controls) and with replication in an additional 2,484 cases and 3,036 controls the authors identified rs3117582, BAT3-MSH5; Pcombined = 4.97 x 10-10) [119]. In a cross-cancer analysis the authors analysed 60,297 single nucleotide polymorphisms, at 229 DNA repair gene regions, using data from the NCI Genetic Associations and Mechanisms in Oncology (GAME-ON) Network. Included were the data from 32 GWAS on 48,734 controls and 51,537 cases across five cancer sites (BC, colon, lung, ovary, and prostate). The authors identified three susceptibility DNA repair genes, RAD51B (P<5.09x10-6), MSH5, rs3115672 (P<5.09x10-6), and BRCA2 (P=5.70x10-6) [120]. SNPs in MSH5 are associated with fewer types of cancer, probably due to lack of relevant studies on this gene.
MSH6 gene (MutS homolog 6)
MSH6 gene is located on chromosome 2 at p16.3 site. There are 15 transcripts of the gene, ten of them are protein-coding. The longest transcript has 12 exons, encoding a 1, 360 amino acid-long protein. The dbSNP database covers 11, 472 SNPs in humans, 48 of them were cited in PubMed. MSH6 heterodimerizes with MSH2 protein to form MutSα complex. Its pivotal role is to recognize base-base mismatches, as well as dinucleotide insertion-deletion loops. When the mismatched region in the DNA is found, it exchanges ADP by ATP to repair DNA mismatch together with MutLα complex [121]. Mutations in the MSH6 gene are frequently associated with HNPCC, CRC and endometrial carcinoma [122].
The most frequently studied SNP of MSH6 is rs1042821 (c.116G>A; Gly39Ala), which was found to have controversial effects in cancer patients. While together with MSH3 rs26279 (Ala1045Thr) it has a protective role in BC patients, alone it acted as a risk factor for BC [73][123]. SNP rs1042821 was associated with decreased risk of the primary hepatocellular cancer, and together with MSH3 rs26279 (Ala1045Thr) with the decreased risk of oesophageal cancer, patients carrying heterozygous G/A genotype of rs1042821 did not display higher risk of CRC (OR=1.65, 95% CI=1.01-2.69, P=0.44) [124][125]. A recent case-control study comprising 106 thyroid cancer patients and 212 age- and gender-matched controls revealed that MSH6 rs1042821 variant homozygotes exhibited higher risk of this cancer (OR=3.42, CI=1.04-11.24, P=0.04). Despite the association was especially evident for the follicular histotype and female sex, the outcomes need to be replicated [126]. Homozygous carriers had a higher risk of CpG island methylated phenotype (CIMP+) in colon cancer (OR=2.20, 95% CI=1.10-4.20) than those with WT genotype [76][124][127[128]. A very significant association with the pancreatic cancer was also found (P ≤ 0.002) for A/G or A/A genotype [129]. A protective effect of exonic region rs1800932 (c.276A>G, Pro92Pro) in CRC onset was observed in the European population comprising 1, 785 cases and 1, 722 control subjects [64]. The intronic SNP rs3136228 (c.-152-405T>G) was identified as a negative marker associated with pronounced adverse effects of chemotherapy. It aggravated the neutropenia in CRC patients treated with FOLFOX4 regimen, decreased the responsiveness to fluoropyrimidine-based chemoradiotherapy in patients with rectal cancer and decreased the OS of prostate cancer patients [68][130][131]. The most significant SNP with adverse effect on cancer risk was rs1042821, associated with pancreatic, prostate and CRC, whereas it had protective roles in the BC, hepatocellular and oesophageal cancers.
PMS1 gene (postmeiotic segregation increased 1)
PMS1 gene is located on chromosome 2 at 2q32.2 locus. The largest mRNA transcript is encoding a 932 amino acid-long protein PMS1, which interacts with MLH1 to form a MutLβ protein complex. PMS1 gene has 22 splicing and 19 protein-coding variants. The longest transcript consists of 12 exons, coding for a protein with 756 amino acids. Up today, 21, 872 SNPs were identified, but only 32 of them were cited in PubMed database. In fact, only a few SNPs in PMS1 are associated with the carcinogenesis, mainly seen in patients with the immunological pathologies. The role of different SNPs in this gene in HNPCC syndrome is still disputed and SNPs are classified as possibly damaging.
Intronic SNP rs5742933 (c.-24G>C) was described as a silencer of exon splicing predicted in a bioinformatic study by PupaSuite software. Based on the PolyPhen scores and availability of three-dimensional structures, structure analysis was carried out with the major mutations that occurred in the native protein [132]. Dong et al. examined 218 SNPs in 50 DNA repair genes in 568 NSCLC patients. Among all those genes, SNP rs5742933 in PMS1 was associated with a worse prognosis (HR=1.89, 95% CI=1.17–3.06, P<0.0001) [133]. Another intronic SNP rs5742938 (c.-21+639G>A) was associated with increased risk of oesophageal cancer in South Africans with mixed ancestry (G/G versus A/A or A/G: OR=1.73, 95% CI=1,07-2.79, P=0.027) but with no effect on the PMS1 mRNA expression levels [127. This variant has never been associated with any cancer except for this study. As it was discussed for MSH2 gene, no exonic SNPs in PMS1 were found to be pathogenic and associated with the increased risk of any cancer. It is disputable why some regions of the human genome are more prone to variations, probably the nucleotide sequence and position on the chromosome play an important role. It is surprising mainly for the MSH2 gene, which is by far the longest MMR one. Only two SNPs were found to have adverse effects in NSCLC and oesophageal cancer development. No protective SNP has been described yet.
PMS2 (postmeiotic segregation increased 1, homolog 2) gene
PMS2 gene is located on chromosome 7 at p22.1 locus. The largest mRNA transcript encodes an 862 amino acid-long protein PMS2, which interacts with the MLH1 to form a MutLα protein complex. Assembly of the MutL-MutS-heteroduplex ternary complex in the presence of the Replication factor C (RFC) and PCNA is sufficient to activate endonuclease activity of PMS2. PMS2 interaction with DNA polymerase III is essential for its recruitment to the site of MMR. The PMS2 gene has 7 splicing variants and the largest one has 15 exons. Up today, 11, 866 SNPs were observed, 39 of them cited in PubMed database. SNPs in PMS2 gene are mainly associated with HNPCC syndrome. For example, seven HNPCC patients were studied for SNP rs1805321 (c.1408C>T, Pro470Ser) in exon 11. Loss of heterozygozity (LOH) was detected in a tumour with the decreased signal from common C allele [134]. Individuals with ovarian cancer and bearing allele A of intronic SNP rs2228006 (c.1621G>A) displayed better OS (HR=0.84, 95% CI=0.71-0.99, P=0.04) [135]. However, the authors concluded the study as probably false positive after adjusting for multiple comparisons. Among 9 SNPs investigated in PMS2 by Li et al., rs2228006 was associated with pancreatic cancer risk (P=0.036) [78]. The intronic SNP rs7797466 (c.24-1121C>T) was associated with a 1.17-fold increase in ovarian cancer risk (95% CI=1.03-1.33, P=0.013) and haplotype analysis showed significant differences in frequencies between cases and controls (P=0.005) [136]. Interestingly, common G/G variant of rs7797466 increased the risk of CRC when combined with G/A variant of MSH6 SNP rs1042821, as investigated in a Polish case-control study of 200 patients and 200 controls (OR=1.65, 95% CI=1.01-2.69, P=0.44) [124]. The SNP rs7797466 was also significantly associated with pancreatic cancer (OR=1.44, 95% CI=1.14-1.81, P≤0.002) on the cohort of 706 patients [129]. Rs7797466 was the most studied SNP of PMS2 and found to be associated with increasing risk of pancreatic, CRC and ovarian cancer. No protective roles of PMS2 SNPs have been described yet.
PMS2P1 (postmeiotic segregation increased 1 homolog 2, pseudogene 1) gene
PMS2P1 gene, also PMS2L1, is located on chromosome 7 at q22.1 site. It is a pseudogene, corresponding to the first 5 exons of PMS2, encoding a 440 amino-acid protein PMS2P1. According to the dbSNP database, 1,445 SNPs were found in human PMS2P1, but none of them was cited in PubMed database or analysed so far. PMS2P1 was characterized by two independent groups and no protein interaction with human MLH1 was observed [137][138][139]. Only one study described the association of a nonsense homozygous mutation in the PMS2P1 gene (c.2428C>T, Arg802X) with a supratentorial primitive neuroectodermal tumour in three siblings, suggesting the involvement of PMS2P1 mutations in childhood cancer [140]. The authors concluded that PMS2P1 mutations in the CRC may be rare, but their involvement in childhood cancers is underestimated. Moreover, previously described polymorphisms in PMS2P1 were, in fact, pseudogene sequence variants of PMS2.
PMS2P2 (postmeiotic segregation increased 1 homolog 2, pseudogene 2) gene
PMS2P2 gene, also PMS2L2, is located on chromosome 7 at q11.23 site. It encodes 297 amino-acid protein PMS2P2, which shares sequence homology with the 5´region of PMS2. PMS2P2 was studied by yeast two-hybrid assay and no interaction with the human MLH1 protein was identified [139]. The authors suggested that PMS2P2 may play a significant role in MMR due to sequence similarity with PMS2. According to the dsSNP database, 1, 299 SNPs were found in human PMS2P2, but none of them was cited in PubMed database or analysed.
5. Role of MMR variants in sporadic cancer
Despite the gaps in understanding functional consequences of SNPs, it has been observed that most of the pathogenic SNPs in MMR genes affect regulatory gene sequences. Mitchell et al. suggested that changes in the regulation of gene expression may have a more significant effect on carcinogenesis than structural changes of the protein product [141]. We may consider at least two molecular mechanisms affecting the regulation of gene expression: changes in gene promoter activity and synthesis of functionally different splicing variants.
One mechanism involved in the regulation of gene expression is represented by promoter methylation. In the majority of sporadic CRC with MSI-high phenotype, MMR system is inactivated mainly by hypermethylation of MLH1 promoter [142]. In this context the possible interference of rs1800734 polymorphisms, residing within the core promoter region of MLH1, with methylation should not be underestimated. The arising errors may alter the length of repetitive DNA sequences (microsatellites) and result in erroneous transcription of tumour suppressor genes and growth factor receptors [5, 6]. This molecular mechanism was also observed in many other malignancies: endometrial and ovarian, laryngeal squamous cell, squamous cell cancer of the lung [143][144][145][146]. Different molecular mechanisms such as allelic imbalance and somatic mutations play probably the minor role in the pathogenesis of CRC. Geisler et al. observed that absent MLH1 expression was associated with the loss of expression of the other five MMR genes (MSH2, MSH3, MSH6, PMS1 and PMS2) [144]. Interestingly, a recent study showed that patients with the HNPCC exhibit the similar cancer prognosis as those with sporadic CRC with confirmed MLH1 promoter hypermethylation [147]. MSH3 promoter hypermethylation was also observed in bladder cancer [47]. The role of SNPs in the regulation of the expression is not clear yet. For example, SNP rs1800734 in the MLH1 gene was found to affect DNA repair capacity in lung cancer patients via transcription regulation [47]. A significant association was shown between SNPs rs1650697 in MSH3 gene and sporadic CRC with MSI in the Japanese population [148]. Two polymorphisms, rs3136229 (c.-448G>A) and rs41540312 (c.-159C>T) in the MSH6 promoter region affected the interaction of Sp1 transcription factor with the MSH6 promoter and increased the sensitivity of the promoter to DNA methylation silencing in vitro [149]. The authors suggested that these SNPs could be preferential sites of the de novo methylation but may also facilitate the binding of other factors to newly methylated DNA, which then amplifies the silencing of MSH6 expression.
The second molecular mechanism affecting the proper function of MMR is a synthesis of different splicing variants due to intronic SNPs. For example, SNP rs2303428 (c.2006-6T>A) in MSH2 intron 12 was studied in meta-analysis for its role in carcinogenesis [150]. The SNP was found to be only associated with the increased risk of non-Hodgkin´s lymphoma (OR=1.62, 95% CI=1.06-2.47). The authors however concluded that this SNP causes a different effect on different types of cancer and further investigation is needed due to lack of data from the reviewed studies and missing studies from African population. One of the first functional studies demonstrated that three SNPs in PMS2 (rs2228007, rs1805318, rs1805324) cause defective protein-protein interaction of PMS2 with MLH1. These SNPs encode the interaction domain for MLH1 [151]. However, the frequencies of these SNPs are below 5 % in the European population.
MMR genes are widely expressed in different types of cells, but organ-specific or tissue-specific mechanisms may occur and become relevant only for some types of cancers. Further investigations are warranted to understand the additional mechanisms by which SNPs in MMR contribute to carcinogenesis in solid tissues. The overview of SNPs with known phenotype is shown in Table 2.
Table 2. The overview of SNPs in EUR population (F≥5 %) with known phenotype in the tumour development: B=benign, D=deleterious, A=ambiguous, P=pathogenic.
|
Gene |
SNP ID |
AA change |
WT-MT |
Ancestral alelle |
Gene region |
Gene position |
Phenotype |
F in EUR pop. |
|
MLH1 |
rs1540354 |
- |
A-T |
T |
intron |
c.307-1403A>T |
B |
16% |
|
|
rs1558528 |
- |
C-A |
A |
intron |
c.67+955C>A |
P |
56% |
|
|
rs2286939 |
- |
T-C |
C |
intron |
c.1038+86T>C |
D |
56% |
|
|
rs4647250 |
- |
T-C |
T |
intron |
c.453+544T>C |
D |
44% |
|
|
rs1799977 |
Ile219Val |
A-G |
A |
Exon 8 |
c.655A>G |
A |
33% |
|
|
rs1800734 |
- |
A-G |
G |
5´ prime |
c.-93G>A |
A |
27% |
|
|
rs2286940 |
- |
C-T |
C |
intron |
c.1410-169C>T |
B |
44% |
|
|
rs9852810 |
- |
G-A |
G |
intron |
c.1410-1306G>A |
P |
44% |
|
|
rs4647269 |
- |
C-T |
C |
intron |
c.791-1406C>T |
A |
44% |
|
MLH3 |
rs108621 |
- |
A-G |
T |
UTR-3 |
c.*3148A>G |
B |
45% |
|
|
rs175080 |
Leu844Pro |
C-T |
G |
Exon 2 |
c.2531C>T |
A |
46% |
|
|
rs28756990 |
Val741Ile |
G-A |
G |
Exon 2 |
c.2221G>A |
P |
5% |
|
|
rs175057 |
- |
G-A |
T |
intron |
c.4012-37G>A |
P |
54% |
|
MSH2 |
rs1981929 |
- |
G-A |
A |
intron |
c.1277-118G>A |
P |
39% |
|
|
rs2042649 |
- |
T-C |
T |
intron |
c.2635-214T>C |
P |
9% |
|
|
rs2059520 |
- |
A-G |
A |
intron |
c.2006-265A>G |
P |
36% |
|
|
rs2303425 |
- |
T-C |
T |
5´prime |
c.-68-50T>C |
P |
15% |
|
|
rs2303426 |
- |
C-G |
G |
intron |
c.211+9C>G |
P |
57% |
|
|
rs2303428 |
- |
T-A |
T |
intron |
c.2006-6T>A |
A |
9% |
|
|
rs3732183 |
- |
G-A |
G |
intron |
c.1661+12G>A |
B |
30% |
|
|
rs6544991 |
- |
A-C |
A |
intron |
c.251+2441A>C |
B |
22% |
|
|
rs10183143 |
- |
T-C |
T |
intron |
c.1661+90T>C |
D |
6% |
|
MSH3 |
rs1650697 |
Ile79Val |
A-G |
C |
Exon 1 |
c.235A>G |
D |
24% |
|
|
rs26279 |
Ala1045Thr |
G-A |
G |
Exon 23 |
c.3133G>A |
A |
72% |
|
|
rs33003 |
- |
A-G |
A |
intron |
c.3303-436A>G |
D |
71% |
|
|
rs184967 |
Gln949Arg |
A-G |
G |
Exon 21 |
c.2846A>G |
D |
14% |
|
|
rs836808 |
- |
G-A |
C |
intron |
c.1174-176G>A |
P |
25% |
|
|
rs863221 |
- |
T-G |
T |
intron |
c.1763+1841T>G |
B |
41% |
|
|
rs1805355 |
Pro231Pro |
G-A |
G |
Exon 4 |
c.693G>A |
P |
5% |
|
|
rs6151627 |
- |
A-G |
A |
intron |
c.580-380A>G |
B |
27% |
|
|
rs1105524 |
- |
A-G |
G |
intron |
c.-35A>G |
P |
32% |
|
|
rs6151670 |
- |
C-G |
C |
intron |
c.1340+8303C>G |
A |
27% |
|
|
rs7709909 |
- |
C-T |
C |
intron |
c.1341-20102C>T |
A |
40% |
|
MSH4 |
rs5745325 |
Ala97Thr |
G-A |
G |
Exon 2 |
c.289G>A |
P |
31% |
|
MSH5 |
rs707938 |
Gln716Gln |
A-G |
C |
Exon 22 |
c.2148A>G |
P |
68% |
|
|
rs2075789 |
Pro29Ser |
C-T |
G |
Exon 2 |
c.85C>T |
D |
10% |
|
|
rs707939 |
- |
C-A |
G |
intron |
c.1326+36C>A |
B |
37% |
|
|
rs3131379 |
- |
G-A |
C |
intron |
c.813-45G>A |
P |
7% |
|
|
rs707939 |
- |
C-A |
G |
intron |
c.1326+36C>A |
A |
37% |
|
MSH6 |
rs1800937 |
Tyr214* |
C-A |
C |
Exon 4 |
c.642C>A |
P |
10% |
|
|
rs1042821 |
Gly39Ala |
G-A |
C |
Exon 1 |
c.116G>A |
A |
18% |
|
|
rs1800935 |
Asp180Asp |
T-C |
T |
Exon 2 |
c.540T>C |
B |
29% |
|
|
rs3136228 |
- |
T-G |
G |
intron |
c.-152-405T>G |
P |
65% |
|
|
rs1800932 |
Pro92Pro |
A-G |
A |
Exon 1 |
c.276A>G |
P |
18% |
|
PMS1 |
rs5742933 |
- |
G-C |
G |
intron |
c.-24G>C |
D |
20% |
|
|
rs5742938 |
- |
G-A |
G |
intron |
c.-21+639G>A |
P |
73% |
|
PMS2 |
rs2228006 |
- |
G-A |
G |
intron |
c.1621G>A |
P |
12% |
|
|
rs7797466 |
- |
C-T |
G |
intron |
c.24-1121C>T |
P |
18% |
6. SNPs in MMR genes and therapy
MMR was indicated to participate in response to many drugs used in anticancer treatment. For example, cytotoxicity of methylating agents (due to O6-methylguanine), is mediated primarily by MMR system components, which recognize mismatches generated by these agents and activate cell cycle arrest and apoptosis [152]. AGEO, a recent large multicentric study, investigated the efficacy of fluoropyrimidine with and without oxaliplatin in a large cohort of MMR-deficient colon cancer patients, since MMR-deficient colon cancer is resistant to 5-fluorouracil adjuvant chemotherapy while preliminary data suggest chemosensitivity to oxaliplatin [153]. Therefore, SNPs in MMR genes may alter the molecular mechanisms of drug response and thus change the responsiveness of patients to treatment. Analysis of SNPs is now considered as an approach helping the oncologist to set a more specific, personalized therapeutic regimen for every single patient and yielding important information for the development of new drugs. Russo et al. supposed that the different responses to a particular drug are not only due to clinicopathological or environmental factors, but also due to the ethnic origins and particular genotype of a patient [154]. Other studies proved that SNPs in MMR genes may substantially change efficacy, toxic effect and the outcome of cancer therapy. For example, SNP rs2303428 in MSH2 was found to be associated with the adverse response and outcome in O6-guanine-based therapy and in radiotherapy. The presence of C allele was significantly overrepresented in acute myeloid leukemia patients previously treated with O6-guanine alkylating agents, compared with controls (OR=4.02, 95% CI=1.40-11.37). [155][156]. The expression of MMR genes may be significantly altered after the chemotherapy, as reported by Murata et al. [153]. The authors observed a strong correlation between pre-surgical chemotherapy and reduced MLH1 expression. They suggested that the silencing may arise due to promoter hypermethylation induced by chemotherapy. Thus, patients carrying SNPs that increase promoter hypermethylation can differently respond to the therapeutic regimen. The reduced expression of MLH1 conferred an advantage for the progression of tumours and hypermethylation of the promoter was linked with clinical stage and lymphatic metastases. Park et al. studied the associations between SNPs and chemotherapy-based treatment of CRC patients. Authors found that SNP rs1625649 in O6-Methylguanine-DNA methyltransferase (MGMT) gene, involved in direct reversal repair, significantly impaired OS of CRC patients treated with oxaliplatin-based therapy, but combined A/G + G/G genotype of the MSH2 rs3732183 (c.1661+12G>A) was associated with better OS (P=0.02) [157]. Other studies on Korean population showed the protective effect of this SNP in CRC and lung cancer. The presence of G/G genotype of rs3732183 was associated with significantly decreased risk of lung adenocarcinoma compared to A/A carriers (N=432 patients, N=432 controls, OR=0.59, 95% CI= 0.40-0.88, P = 0.01). The CRC patients with the combined G/G+A/G genotype revealed better OS than patients with A/A genotype (N=379 cancer patients, HR=0.50, 95% CI=0.26–0.98, P=0.042) [158][159]. Another SNP rs6544991 (c.*251+2441A>C) in MSH2 gene was associated with the increased toxicity of platinum-based chemotherapy within NSCLC treatment [116]. This study demonstrated that the rs6544991 C/C genotype was associated with gastrointestinal toxicity, MSH3 rs6151627 (c.580-380A>G), rs6151670 (c.1340+8303C>G), and rs7709909 (c.1341-20102C>T) were associated with hematologic toxicity, and MSH5 rs707939 (c.1326+36C>A) was associated with gastrointestinal and overall toxicity. The other study focused on the role of SNPs in MMR genes (MLH1, MSH2, MGMT) in melanoma patients after temozolomide and dacarbazine treatment, including comparison with five melanoma cell lines [160]. The authors observed the decreased expression of the MLH1 gene in patients bearing SNP rs1799977 and found five new SNPs in MLH1. The presence of SNPs and promoter methylation of MMR genes was identified as a major cause of melanoma treatment resistance. The role of rs1799977 in MLH1 gene was also studied in B-cell lymphoma patients, treated with R-CHOP21 (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisolone administered every 21 days) and platinum-based therapy. Patients carrying either A/G or G/G genotype showed an increased risk of death (HR=3.21, P<0.001) compared to those carrying the A/A genotype [63]. This study vindicated that MMR components can significantly regulate the genotoxicity of doxorubicin and platinum-based therapy. The role of variants in MMR genes in CRC development was studied in the Czech population, comprising 1, 095 cases and 1, 469 controls [15]. Carriers of the C/C genotype of rs108621 (c.*3148A>G) in the 3'UTR of MLH3 (a microRNA binding site), after 5-fluorouracil-based (5-FU) chemotherapy, showed a significantly increased OS compared to those with either C/T or T/T genotype (P=0.05). Moreover, heterozygous carriers had a higher risk of relapse (P=0.03). Patients carrying the C/C genotype for MSH6 rs1800935 (Asp180Asp) and not undergoing 5-FU-based chemotherapy showed a decreased risk of recurrences (P=0.03). In another study the same authors investigated the epigenetic mechanisms and the expression of MMR genes comparing the tumours and adjacent mucosa tissues in sporadic CRC patients. They observed different expression according to tumour localization - colon tumours had higher expression of MMR genes in comparison to rectal cancer (P=0.02). Only 9 % of tumours were positive for the methylation of the MLH1 gene [161]. The SNP rs3136228 in MSH6 was studied for its contribution to CRC development. A prospective study in CRC patients treated with FOLFOX4 regimen revealed possible association of rs3136228 in MSH6 with grade 3 neutropenia toxicity (P=0.07). This SNP may affect the MMR activity in non-malignant cells and therefore modulate the genotoxic effect of FOLFOX therapy [131]. The same group showed that this SNP is in the upstream region of the gene and seems to affect the binding with the transcription factor Sp1, followed by decreased expression of MSH6 and thus MMR efficiency [68]. The strongest association (P<0.01) between genetic variants and pathological response to neoadjuvant treatment of rectal cancer was observed particularly for rs3136228 in MSH6 gene in the subgroup of patients undergoing standard treatment-5-FU combined with a dose of 5,040 cGy of radiotherapy. Other SNPs in 21 genes did not show any significant association. The overview of SNPs evaluated in clinical studies is presented in Table 3.
Table 3: The overview of SNPs studied for their role in the therapy of various types of tumours.
|
Gene |
SNP ID |
PubMed ID |
Reference |
Cancer |
Cases |
Controls |
Phenotype |
|
MLH1 |
rs1799977 |
19203531 |
160 |
Melanoma |
51 |
0 |
- |
|
rs1799977 |
21156845 |
63 |
B-cell lymph. |
308 |
0 |
↓ effect of Dox treatment in folicullar lymphoma |
|
|
rs1799977 |
21156845 |
63 |
B-cell lymph. |
308 |
0 |
↓ effect of Pt-based second line treatment |
|
|
rs1799977 |
27608007 |
68 |
Rectal |
280 |
0 |
protective factor in rectal c. |
|
|
rs1800734 |
25047469 |
|
SCC |
185 |
0 |
GA genotype ↓ OS in oral squamous cell c. |
|
|
rs1800734 |
25047469 |
|
SCC |
185 |
0 |
AA genotype ↓ OS in oral squamous cell c. |
|
|
rs2286940 |
26743341 |
70 |
Lymphoid |
153 |
0 |
sign. predictor of progression-free surv. |
|
|
MLH3 |
rs108621 |
24755277 |
15 |
CRC |
1095 |
1469 |
CC genotype ↑ survival |
|
MSH2 |
rs2042649 |
22017238 |
|
Glioblastoma |
121 |
0 |
↑ OS of glioblastoma patients |
|
rs2303428 |
22017238 |
|
Glioblastoma |
121 |
0 |
↑ OS of glioblastoma patients |
|
|
rs2303428 |
19741564 |
|
Melanoma |
51 |
0 |
↑ hematologic side effects in melanoma tr. |
|
|
rs2303428 |
20458443 |
|
NSCLC |
96 |
0 |
CC genotype ↑ response to Pt-based therapy |
|
|
rs2303428 |
20708344 |
|
Breast |
87 |
0 |
↑ radiosensitivity in breast c. patients |
|
|
rs3732183 |
20091185 |
157 |
CRC |
94 |
0 |
AG/GG genotype ↑ response to therapy |
|
|
rs6544991 |
28093084 |
116 |
NSCLC |
220 |
0 |
↑ GIT toxicity of Pt-based chemo |
|
|
MSH3 |
rs863221 |
19115210 |
95 |
CRC |
2060 |
0 |
↑ OS in patients with CRC |
|
rs26279 |
25966119 |
97 |
NSCLC |
180 |
0 |
↑ sensitivity to Pt-based therapy in NSCLC |
|
|
rs6151627 |
28093084 |
116 |
NSCLC |
220 |
0 |
↑ hematological toxicity in NSCLC patients |
|
|
rs1105524 |
25966119 |
97 |
NSCLC |
180 |
0 |
G/A and A/A genotype ↓ OS in NSCLC |
|
|
MSH5 |
rs707939 |
28093084 |
116 |
NSCLC |
220 |
0 |
↑ hematological toxicity in NSCLC patients |
|
MSH6 |
rs3136228 |
22868256 |
131 |
CRC |
144 |
0 |
grade 3-4 neutropenia |
|
rs3136228 |
27608007 |
68 |
Rectal |
280 |
0 |
↓ response to F-pyrimidine therapy |
|
|
rs3136228 |
28206966 |
130 |
Prostate |
542 |
0 |
↓ OS in prostate c. patients |
|
|
rs1800935 |
24755277 |
15 |
CRC |
1095 |
1469 |
CC carriers had ↓ recurrency of CRC |
7. Conclusions
The overall conclusion is that the role of SNPs in the MMR pathway in carcinogenesis is not clear yet. Available publications reveal substantial variability among SNPs in various types of cancer. Some genetic variants have a beneficial effect in a particular cancer, some of them are pathological or often described as possibly damaging. The pool of available studies contains reports of various size and quality and the studies based on a limited number of patients may bias the overall outcome. The manuscripts considered in our review disclose for the first time the implication of same variants in MMR genes (such as in MLH1, MSH2, MSH3, and MSH6) in the risk of different cancers. These SNPs will deserve attention in the future studies.
One of the main problems is a lack of functional studies which may provide evidence of the function and ultimate pathogenicity of a particular SNP. Apparently, most of the pathogenic SNPs in the group of MMR genes affect regulatory sequences, important for proper gene expression. Therefore, it seems that these changes are more or equally involved in the carcinogenesis than structural changes in the relevant protein product. Additional aspect, worthy of consideration, is tumour heterogeneity. According to some GWAS studies, SNPs correlate with the tumour stage or grade. In general, the authors of GWAS studies did not focus on the possible mechanism by which the particular SNP modulated the function of MMR gene.
Results from all available studies suggest that single SNPs in MMR genes may contribute to sporadic cancer susceptibility, cancer progression and may affect the response to therapy. In our recent review, we noted that individual genetic variation in biotransformation, DNA repair and mitosis regulating genes exerted small modulating effects on chromosomal aberrations, in interactions their effect was more pronounced [162]. The same may apply to the role of MMR gene variants in complex diseases, such as cancer. The interactions (mainly binary) between MMR SNPs are only fragmentarily addressed on relatively small studies. Further research in this direction may provide important outcomes.
Extensive interactions between proteins of distinct DNA repair pathways need to be considered in evaluating the DDR-related risk as well [30], understanding these interplays may serve as prognostic and predictive factors for several malignancies.
References
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Bien, S.; Zubair, N. Genetic architecture of colorectal cancer. Gut 2015, 64, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2014, 60, 762–772. [Google Scholar] [CrossRef]
- Nielsen, F.C.; Hansen, T.V.O.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Niskakoski, A.; Pasanen, A.; Lassus, H.; Renkonen-Sinisalo, L.; Kaur, S.; Mecklin, J.-P.; Bützow, R.; Peltomäki, P. Molecular changes preceding endometrial and ovarian cancer: A study of consecutive endometrial specimens from Lynch syndrome surveillance. Mod. Pathol. 2018, 31, 1291–1301. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R. Genetic Diagnosis of High-Penetrance Susceptibility for Colorectal Cancer (CRC) Is Achievable for a High Proportion of Familial CRC by Exome Sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef]
- Peltomäki, P. Role of DNA Mismatch Repair Defects in the Pathogenesis of Human Cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; De Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Lavebratt, C.; Şengül, S. Single nucleotide polymorphism (SNP) allele frequency estimation in DNA pools using Pyrosequencing™. Nat. Protoc. 2006, 1, 2573–2582. [Google Scholar] [CrossRef]
- Mohrenweiser, H.W.; Xi, T.; Vázquez-Matías, J.; Jones, I.M. Identification of 127 amino acid substitution variants in screening 37 DNA repair genes in humans. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1054–1064. [Google Scholar]
- Vymetalkova, V.; Pardini, B.; Rosa, F.; Di Gaetano, C.; Novotny, J.; Levy, M.; Büchler, T.; Slyskova, J.; Vodickova, L.; Naccarati, A.; et al. Variations in mismatch repair genes and colorectal cancer risk and clinical outcome. Mutagenesis 2014, 29, 259–265. [Google Scholar] [CrossRef]
- Han, W.; Kim, K.-Y.; Yang, S.-J.; Noh, D.-Y.; Kang, D.; Kwack, K. SNP-SNP interactions between DNA repair genes were associated with breast cancer risk in a Korean population. Cancer 2011, 118, 594–602. [Google Scholar] [CrossRef]
- Schwender, H.R.; Selinski, S.; Blaszkewicz, M.; Marchan, R.; Ickstadt, K.; Golka, K.; Hengstler, J.G. Distinct SNP Combinations Confer Susceptibility to Urinary Bladder Cancer in Smokers and Non-Smokers. PLoS ONE 2012, 7, e51880. [Google Scholar] [CrossRef]
- Ito, H.; Sueta, A.; Iwata, H.; Hosono, S.; Oze, I.; Watanabe, M.; Iwase, H.; Tanaka, H.; Matsuo, K. Abstract 06: A genetic risk predictor for breast cancer using a combination of low-penetrance polymorphisms in a Japanese population. Cancer Epidemiol. Biomark. Prev. 2012, 21, 6. [Google Scholar] [CrossRef]
- Bhushan, S.; McLeod, H.; Walko, C.M. Role of Pharmacogenetics as Predictive Biomarkers of Response and/or Toxicity in the Treatment of Colorectal Cancer. Clin. Color. Cancer 2009, 8, 15–21. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef]
- Lahue, R.S.; Au, K.G.; Modrich, P. DNA mismatch correction in a defined system. Science 1989, 245, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Nevers, P.; Spatz, H.C. Escherichia coli mutants uvr D and uvr E deficient in gene conversion of lambda-heteroduplexes. Mol. Gen. Genet. 1975, 139, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Ban, C.; Junop, M.; Yang, W. Transformation of MutL by ATP binding and hydrolysis: A switch in DNA mismatch repair. Cell 1999, 97, 85–97. [Google Scholar] [CrossRef]
- Längle-Rouault, F.; Maenhaut-Michel, G.; Radman, M. GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch repair. EMBO J. 1987, 6, 1121–1127. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLα in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef]
- Genschel, J.; Littman, S.J.; Drummond, J.T.; Modrich, P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J. Biol. Chem. 1998, 273, 19895–19901. [Google Scholar] [CrossRef]
- Sharma, M.; Predeus, A.V.; Kovacs, N.; Feig, M. Differential Mismatch Recognition Specificities of Eukaryotic MutS Homologs, MutSα and MutSβ. Biophys. J. 2014, 106, 2483–2492. [Google Scholar] [CrossRef]
- Marsischky, G.T.; Filosi, N.; Kane, M.F.; Kolodner, R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996, 10, 407–420. [Google Scholar] [CrossRef]
- Gupta, S.; Gellert, M.; Yang, W. Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2011, 19, 8–72. [Google Scholar]
- Kondo, E. The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS. Nucleic Acids Res. 2001, 29, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Banasik, M.; Sachadyn, P. Conserved motifs of MutL proteins. Mutat. Res. Mol. Mech. Mutagenesis 2014, 769, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Musak, L.; Frank, C.; Kazimirova, A.; Vymetalkova, V.; Barancokova, M.; Smolkova, B.; Dzupinkova, Z.; Jiraskova, K.; Vodenkova, S.; et al. Interactions of DNA repair gene variants modulate chromosomal aberrations in healthy subjects. Carcinogenesis 2015, 36, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Larrea, A.A.; Lujan, S.A.; Kunkel, T.A. SnapShot: DNA Mismatch Repair. Cell 2010, 141, 730–730.e1. [Google Scholar] [CrossRef]
- Peltomäki, P.; Vasen, H. Mutations predisposing to hereditary nonpolyposis colorectal cancer: Database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 1997, 113, 1146–1158. [Google Scholar] [CrossRef]
- Mrkonjic, M.; Roslin, N.M.; Greenwood, C.M.T.; Raptis, S.; Pollett, A.; Laird, P.W.; Pethe, V.V.; Chiang, T.; Daftary, D.; Dicks, E.; et al. Specific Variants in the MLH1 Gene Region May Drive DNA Methylation, Loss of Protein Expression, and MSI-H Colorectal Cancer. PLoS ONE 2010, 5, e13314. [Google Scholar] [CrossRef]
- Nizam, Z.M.; Aziz, A.A.A.; Kaur, G.; Abu Hassan, M.R.; Sidek, A.S.M.; Lee, Y.Y.; Mazuwin, M.; Ankathil, R. Contribution of the MLH1 −93G>A Promoter Polymorphism in Modulating Susceptibility Risk in Malaysian Colorectal Cancer Patients. Asian Pac. J. Cancer Prev. 2013, 14, 619–624. [Google Scholar] [CrossRef]
- Beiner, M.E.; Rosen, B.; Harley, I.; Siminovitch, K.; Zhang, S.; Fyles, A.; Pal, T.; Sun, P.; Narod, S.A. Endometrial Cancer Risk Is Associated with Variants of the Mismatch Repair Genes MLH1 and MSH. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1636–1640. [Google Scholar] [CrossRef]
- Whiffin, N.; Broderick, P.; Lubbe, S.J.; Pittman, A.; Penegar, S.; Chandler, I.; Houlston, R.S. MLH1-93G > A is a risk factor for MSI colorectal cancer. Carcinogenesis 2011, 32, 1157–1161. [Google Scholar] [CrossRef]
- Koref, M.F.S.; Wilson, V.; Cartwright, N.; Cunnington, M.S.; Mathers, J.C.; Bishop, D.; Curtis, A.; Dunlop, M.G.; Burn, J. MLH1 Differential Allelic Expression in Mutation Carriers and Controls. Ann. Hum. Genet. 2010, 74, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.; Houlston, R.S.; Montgomery, G.W.; Sieber, O.; Dunlop, M.G. Investigation of the effects of DNA repair gene polymorphisms on the risk of colorectal cancer. Mutagenesis 2012, 27, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.M.; Shorto, J.; Adlard, J.; Bury, J.; Coggins, R.; George, R.; Katory, M.; Quirke, P.; Richman, S.; Scott, D.; et al. MLH1 −93G>A promoter polymorphism and risk of mismatch repair deficient colorectal cancer. Int. J. Cancer 2008, 123, 2456–2459. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, I.; Perdomo, S.; Santos-Briz, A.; Hernández, J.L.G.; Gómez-Moreta, J.A.; Cruz, J.J.; González-Sarmiento, R. Analysis of DNA repair gene polymorphisms in glioblastoma. Gene 2014, 536, 79–83. [Google Scholar] [CrossRef]
- Lo, Y.-L.; Hsiao, C.-F.; Jou, Y.-S.; Chang, G.-C.; Tsai, Y.-H.; Su, W.-C.; Chen, K.-Y.; Chen, Y.-M.; Huang, M.-S.; Hsieh, W.-S.; et al. Polymorphisms of MLH1 and MSH2 genes and the risk of lung cancer among never smokers. Lung Cancer 2011, 72, 280–286. [Google Scholar] [CrossRef]
- Shih, C.-M.; Chen, C.-Y.; Lee, I.-H.; Kao, W.-T.; Wang, Y.-C. A polymorphism in the hMLH1 gene (−93G-->A) associated with lung cancer susceptibility and prognosis. Int. J. Mol. Med. 2010, 25, 165–170. [Google Scholar]
- Park, S.H.; Lee, G.Y.; Jeon, H.-S.; Lee, S.J.; Kim, K.M.; Jang, S.S.; Kim, C.H.; Lee, W.K.; Kam, S.; Park, R.W.; et al. −93G→A polymorphism ofhMLH1 and risk of primary lung cancer. Int. J. Cancer 2004, 112, 678–682. [Google Scholar] [CrossRef]
- Halasova, E.; Matáková, T.; Skerenova, M.; Krutakova, M.; Slovakova, P.; Dzian, A.; Javorkova, S.; Pec, M.; Kypusova, K.; Hamzik, J. Polymorphisms of Selected DNA Repair Genes and Lung Cancer in Chromium Exposure. Retin. Degener. Dis. 2016, 911, 17–22. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Yuan, S.; Xie, W.; Liu, Y.; Xiang, Y.; Wu, N.; Wu, L.; Ma, X.; Cai, T.; et al. Genetic predisposition to lung cancer: Comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Raptis, S.; Mrkonjic, M.; Green, R.C.; Pethe, V.V.; Monga, N.; Chan, Y.M.; Daftary, D.; Dicks, E.; Younghusband, B.H.; Parfrey, P.S.; et al. MLH1 −93G>A Promoter Polymorphism and the Risk of Microsatellite-Unstable Colorectal Cancer. J. Natl. Cancer Inst. 2007, 99, 463–474. [Google Scholar] [CrossRef]
- Liu, N.Q.; Ter Huurne, M.; Nguyen, L.N.; Peng, T.; Wang, S.-Y.; Studd, J.B.; Joshi, O.; Ongen, H.; Bramsen, J.B.; Yan, J.; et al. The non-coding variant rs1800734 enhances DCLK3 expression through long-range interaction and promotes colorectal cancer progression. Nat. Commun. 2017, 8, 14418. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Mrkonjic, M.; Rawson, J.R.; Bapatet, B. Functional effects of the MLH1-93G>A polymorphism on MLH1/EPM2AIP1 promoter activity. Oncol. Rep. 2011, 25, 809–815. [Google Scholar]
- Pan, X.-M.; Yang, W.-Z.; Xu, G.-H.; Bai, P.; Qin, H.-J.; Zhang, L.-S.; Zhai, X.-D.; Tang, M.; Deng, W.; Gao, L.-B. The association between MLH1 −93 G>A polymorphism of DNA mismatch repair and cancer susceptibility: A meta-analysis. Mutagenesis 2011, 26, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Tulupova, E.; Kumar, R.; Hanova, M.; Slyskova, J.; Pardini, B.; Polakova, V.; Naccarati, A.; Vodickova, L.; Novotny, J.; Halamkova, J.; et al. Do polymorphisms and haplotypes of mismatch repair genes modulate risk of sporadic colorectal cancer. Mutat. Res. Mol. Mech. Mutagen. 2008, 648, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Corrado, A.; Paolicchi, E.; Cugliari, G.; Berndt, S.I.; Bezieau, S.; Bien, S.A.; Brenner, H.; Caan, B.J.; Campbell, P.T.; et al. DNA repair and cancer in colon and rectum: Novel players in genetic susceptibility. Int. J. Cancer 2019, 146, 363–372. [Google Scholar] [CrossRef]
- Cervena, K.; Siskova, A.; Buchler, T.; Vodicka, P.; Vymetalkova, V. Methylation-Based Therapies for Colorectal Cancer. Cells 2020, 9, 1540. [Google Scholar] [CrossRef]
- Savio, A.J.; Lemire, M.; Mrkonjic, M.; Gallinger, S.; Zanke, B.W.; Hudson, T.J.; Bapat, B. MLH1 Region Polymorphisms Show a Significant Association with CpG Island Shore Methylation in a Large Cohort of Healthy Individuals. PLoS ONE 2012, 7, e51531. [Google Scholar] [CrossRef]
- Liu, B.; Nicolaides, N.C.; Markowitz, S.; Willson, J.K.V.; Parsons, R.; Jen, J.; Papadopolous, N.; Peltomäki, P.; De La Chapelle, A.; Hamilton, S.R.; et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat. Genet. 1995, 9, 48–55. [Google Scholar] [CrossRef]
- Nejda, N.; Iglesias, D.; Azcoita, M.M.; Arana, V.M.; González-Aguilera, J.J.; Fernández-Peralta, A.M. A MLH1 polymorphism that increases cancer risk is associated with better outcome in sporadic colorectal cancer. Cancer Genet. Cytogenet. 2009, 193, 71–77. [Google Scholar] [CrossRef]
- Smith, T.R.; Levine, E.A.; Freimanis, R.I.; Akman, S.A.; Allen, G.O.; Hoang, K.N.; Liu-Mares, W.; Hu, J.J. Polygenic model of DNA repair genetic polymorphisms in human breast cancer risk. Carcinogenesis 2008, 29, 2132–2138. [Google Scholar] [CrossRef]
- An, Y.; Jin, G.; Wang, H.; Liu, H.; Li, R.; Wang, H.; Qian, J.; Sun, W.; Wang, Y.; Ma, H.; et al. Polymorphisms in hMLH1 and risk of early-onset lung cancer in a southeast Chinese population. Lung Cancer 2008, 59, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, W.J.; Kwon, E.M.; Koopmeiners, J.S.; Ostrander, E.A.; Stanford, J.L. Population-based study of the association of variants in mismatch repair genes with prostate cancer risk and outcomes. Cancer Epidemiol. Biomark. Prev. 2010, 19, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Di Rocco, A.; Fabbri, A.; Forconi, F.; Gloghini, A.; Bruscaggin, A.; Franceschetti, S.; Fangazio, M.; De Paoli, L.; et al. The host genetic background of DNA repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood 2011, 117, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Zajac, P.; Zhou, X.-L.; Edler, D.; Lenander, C.; Dalén, J.; Hjern, F.; Lundqvist, N.; Lindforss, U.; Påhlman, L.; et al. Common variants in human CRC genes as low-risk alleles. Eur. J. Cancer 2010, 46, 1041–1048. [Google Scholar] [CrossRef]
- Landi, S.; Gemignani, F.; Canzian, F.; Gaborieau, V.; Barale, R.; Landi, D.; Szeszenia-Dabrowska, N.; Zaridze, D.G.; Lissowska, J.; Rudnai, P.; et al. DNA Repair and Cell Cycle Control Genes and the Risk of Young-Onset Lung Cancer. Cancer Res. 2006, 66, 11062–11069. [Google Scholar] [CrossRef]
- Campbell, P.T.; Curtin, K.; Ulrich, C.M.; Samowitz, W.S.; Bigler, J.; Velicer, C.M.; Caan, B.; Potter, J.D.; Slattery, M.L. Mismatch repair polymorphisms and risk of colon cancer, tumour microsatellite instability and interactions with lifestyle factors. Gut 2008, 58, 661–667. [Google Scholar] [CrossRef]
- Picelli, S.; Bermejo, J.L.; Chang-Claude, J.; Hoffmeister, M.; Fernandez-Rozadilla, C.; Carracedo, A.; Castells, A.; Castellvi-Bel, S.; Naccarati, A.; Pardini, B.; et al. Meta-Analysis of Mismatch Repair Polymorphisms within the Cogent Consortium for Colorectal Cancer Susceptibility. PLoS ONE 2013, 8, e72091. [Google Scholar] [CrossRef]
- Dreussi, E.; Cecchin, E.; Polesel, J.; Canzonieri, V.; Agostini, M.; Boso, C.; Belluco, C.; Buonadonna, A.; Lonardi, S.; Bergamo, F.; et al. Pharmacogenetics Biomarkers and Their Specific Role in Neoadjuvant Chemoradiotherapy Treatments: An Exploratory Study on Rectal Cancer Patients. Int. J. Mol. Sci. 2016, 17, 1482. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Hu, F.; Bi, H.; Wu, Z.; Zhao, X.; Li, Y.; Li, S.; Li, D.; Cui, B.; et al. The prognostic significance of polymorphisms in hMLH1/hMSH2 for colorectal cancer. Med. Oncol. 2014, 31, 975. [Google Scholar] [CrossRef]
- Shinozuka, K.; Tang, H.; Jones, R.B.; Li, D.; Nieto, Y. Impact of Polymorphic Variations of Gemcitabine Metabolism, DNA Damage Repair, and Drug-Resistance Genes on the Effect of High-Dose Chemotherapy for Relapsed or Refractory Lymphoid Malignancies. Biol. Blood Marrow Transplant. 2016, 22, 843–849. [Google Scholar] [CrossRef]
- Sapkota, Y.; Mackey, J.R.; Lai, R.; Franco-Villalobos, C.; Lupichuk, S.; Robson, P.; Kopciuk, K.; Cass, C.E.; Yasui, Y.; Damaraju, S. Assessing SNP-SNP Interactions among DNA Repair, Modification and Metabolism Related Pathway Genes in Breast Cancer Susceptibility. PLoS ONE 2013, 8, e64896. [Google Scholar] [CrossRef] [PubMed]
- Kan, R.; Sun, X.; Kolas, N.K.; Avdievich, E.; Kneitz, B.; Edelmann, W.; Cohen, P.E. Comparative Analysis of Meiotic Progression in Female Mice Bearing Mutations in Genes of the DNA Mismatch Repair Pathway. Biol. Reprod. 2008, 78, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Silva, S.N.; Azevedo, A.P.; Teixeira, V.; Pina, J.E.; Rueff, J.; Gaspar, J. Association of common variants in mismatch repair genes and breast cancer susceptibility: A multigene study. BMC Cancer 2009, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Santucci-Darmanin, S.; Neyton, S.; Lespinasse, F.; Saunières, A.; Gaudray, P.; Paquis-Flucklinger, V. The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum. Mol. Genet. 2002, 11, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Michiels, S.; Danoy, P.; Dessen, P.; Bera, A.; Boulet, T.; Bouchardy, C.; Lathrop, M.; Sarasin, A.; Benhamou, S. Polymorphism discovery in 62 DNA repair genes and haplotype associations with risks for lung and head and neck cancers. Carcinogenesis 2007, 28, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Jia, J.; Tang, L.; Gao, X.; Yan, L.; Wang, L.; Yu, F.; Ma, N.; Liu, W.; et al. Correlation between polymorphisms in DNA mismatch repair genes and the risk of primary hepatocellular carcinoma for the Han population in northern China. Scand. J. Gastroenterol. 2015, 50, 1–7. [Google Scholar] [CrossRef]
- Ye, F.; Cheng, Q.; Shen, J.; Zhou, C.; Chen, H. Mismatch Repair Gene MLH3 Pro844Leu and Thr942Ile Polymorphisms and the Susceptibility to Cervical Carcinoma and HPV Infection: A Case-Control Study in a Chinese Population. PLoS ONE 2014, 9, e96224. [Google Scholar] [CrossRef]
- Li, N.; Duell, E.J.; Yu, K.; Risch, H.A.; Olson, S.H.; Kooperberg, C.; Wolpin, B.M.; Jiao, L.; Dong, X.; Wheeler, B.; et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis 2012, 33, 1384–1390. [Google Scholar] [CrossRef]
- Lin, X.; Chen, Z.; Gao, P.; Gao, Z.; Chen, H.; Qi, J.; Liu, F.; Ye, D.; Jiang, H.; Na, R.; et al. TEX15: A DNA repair gene associated with prostate cancer risk in Han Chinese. Prostate 2017, 77, 1271–1278. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, R.; Wang, X.W.; Linke, S.P.; Sengupta, S.; Hickson, I.D.; Pedrazzi, G.; Perrera, C.; Stagljar, I.; Littman, S.J.; et al. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene 2004, 23, 3749–3756. [Google Scholar] [CrossRef]
- Seifert, M.; Scherer, S.J.; Edelmann, W.; Böhm, M.; Meineke, V.; Löbrich, M.; Tilgen, W.; Reichrath, J. The DNA-Mismatch Repair Enzyme hMSH2 Modulates UV-B-Induced Cell Cycle Arrest and Apoptosis in Melanoma Cells. J. Investig. Dermatol. 2008, 128, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Slováková, P.; Majerová, L.; Matakova, T.; Skerenova, M.; Kavcová, E.; Halasova, E. Mismatch Repair Gene Polymorphisms and Association with Lung Cancer Development. Retin. Degener. Dis. 2014, 833, 15–22. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Cho, E.-C.; Tu, S.-H.; Wu, C.-H.; Hung, C.-S.; Hsieh, M.-C.; Su, C.-T.; Liu, Y.-R.; Lee, C.-H.; Ho, Y.-S.; et al. MSH2 rs2303425 Polymorphism is Associated with Early-Onset Breast Cancer in Taiwan. Ann. Surg. Oncol. 2016, 24, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Mrkonjic, M.; Raptis, S.; Green, R.C.; Monga, N.; Daftary, D.; Dicks, E.; Younghusband, H.; Parfrey, P.S.; Gallinger, S.S.; McLaughlin, J.R.; et al. MSH2 −118T>C and MSH6 −159C>T promoter polymorphisms and the risk of colorectal cancer. Carcinogenesis 2007, 28, 2575–2580. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Srivastava, A.; Kumar, A.; Mittal, B. Gallbladder Cancer Predisposition: A Multigenic Approach to DNA-Repair, Apoptotic and Inflammatory Pathway Genes. PLoS ONE 2011, 6, e16449. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, G.A.S.; Lourenço, G.J.; Oliveira, C.B.M.; Marson, F.A.L.; Lopes-Aguiar, L.; Costa, E.F.D.; Lima, T.R.P.; Liutti, V.; Leal, F.; Santos, V.C.A.; et al. Association between genetic polymorphisms in DNA mismatch repair-related genes with risk and prognosis of head and neck squamous cell carcinoma. Int. J. Cancer 2015, 137, 810–818. [Google Scholar] [CrossRef]
- Sanguansin, S.; Petmitr, S.; Punyarit, P.; Vorasubin, V.; Weerapradist, W.; Surarit, R. HMSH2 gene alterations associated with recurrence of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2006, 25, 251–257. [Google Scholar]
- Wang, D.; Zhou, J.; Wang, T.; Li, X.; Li, S.; Chen, S.; Ma, G.; Li, J.; Zhang, X. Polymorphisms in MSH2 gene and risk of gastric cancer, and interactions with lifestyle factors in a Chinese population. Cancer Epidemiol. 2012, 36, e171–e176. [Google Scholar] [CrossRef]
- Park, J.M.; Huang, S.; Tougeron, D.; Sinicrope, F.A. MSH3 Mismatch Repair Protein Regulates Sensitivity to Cytotoxic Drugs and a Histone Deacetylase Inhibitor in Human Colon Carcinoma Cells. PLoS ONE 2013, 8, e65369. [Google Scholar] [CrossRef]
- Kleczkowska, H.E.; Marra, G.; Lettieri, T.; Jiricny, J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001, 15, 724–736. [Google Scholar] [CrossRef]
- Marra, G.; Iaccarino, I.; Lettieri, T.; Roscilli, G.; Delmastro, P.; Jiricny, J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl. Acad. Sci. USA 1998, 95, 8568–8573. [Google Scholar] [CrossRef] [PubMed]
- Berndt, S.I.; Platz, E.A.; Fallin, M.D.; Thuita, L.W.; Hoffman, S.C.; Helzlsouer, K.J. Mismatch repair polymorphisms and the risk of colorectal cancer. Int. J. Cancer 2007, 120, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Koessler, T.; Oestergaard, M.Z.; Tyrer, J.; Perkins, B.; Dunning, A.M.; Pharoah, P.D.P.; Song, H.; Easton, D.F. Common variants in mismatch repair genes and risk of colorectal cancer. Gut 2008, 57, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.-K.; Chen, L.-P.; Cai, D.-P.; Kong, W.-J.; Xiao, L.; Lin, J. MSH3 rs26279 polymorphism increases cancer risk: A meta-analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 11060–11067. [Google Scholar] [PubMed]
- Koessler, T.; Azzato, E.M.; Perkins, B.; Maclnnis, R.; Greenberg, D.; Easton, U.F.; Pharoah, P.D. Common germline variation in mismatch repair genes and survival after a diagnosis of colorectal cancer. Int. J. Cancer 2009, 124, 1887–1891. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Herranz, J.; Michailidou, K.; Dennis, J.; Tyrer, J.P.; Zamora, M.P.; Perez, J.I.A.; González-Neira, A.; Pita, G.; Alonso, M.R.; et al. A large-scale assessment of two-way SNP interactions in breast cancer susceptibility using 46 450 cases and 42 461 controls from the breast cancer association consortium. Hum. Mol. Genet. 2013, 23, 1934–1946. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-L.; Yao, Y.-L.; Xu, W.-Z.; Feng, J.-G.; Mao, W.-M. Correlation of MSH3 polymorphisms with response and survival in advanced non-small cell lung cancer patients treated with first-line platinum-based chemotherapy. Genet. Mol. Res. 2015, 14, 3525–3533. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Daley, D.; Leach, S.; Conde, L.; Berry, B.R.; Gallagher, R.P.; Connors, J.M.; Gascoyne, R.D.; Bracci, P.M.; Skibola, C.F.; et al. Non-Hodgkin Lymphoma Risk and Variants in Genes Controlling Lymphocyte Development. PLoS ONE 2013, 8, e75170. [Google Scholar] [CrossRef]
- Skibola, C.F.; Bracci, P.M.; Halperin, E.; Nieters, A.; Hubbard, A.; Paynter, R.A.; Skibola, D.R.; Agana, L.; Becker, N.; Tressler, P.; et al. Polymorphisms in the Estrogen Receptor 1 and Vitamin C and Matrix Metalloproteinase Gene Families Are Associated with Susceptibility to Lymphoma. PLoS ONE 2008, 3, e2816. [Google Scholar] [CrossRef]
- Paquis-Flucklinger, V.; Santucci-Darmanin, S.; Paul, R.; Saunières, A.; Turc-Carel, C.; Desnuelle, C. Cloning and Expression Analysis of a Meiosis-Specific MutS Homolog: The HumanMSH4Gene. Genomics 1997, 44, 188–194. [Google Scholar] [CrossRef]
- Pochart, P.; Woltering, D.; Hollingsworth, N.M. Conserved Properties between Functionally Distinct MutS Homologs in Yeast. J. Biol. Chem. 1997, 272, 30345–30349. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; MacQueen, A.J.; Duffy, J.B.; Kemphues, K.J.; Villeneuve, A.M. Crossing over during Caenorhabditis elegans meiosis requires a conserved MutS-based pathway that is partially dispensable in budding yeast. Genetics 1999, 153, 1271–1283. [Google Scholar] [PubMed]
- Bocker, T.; Barusevicius, A.; Snowden, T.; Rasio, D.; Guerrette, S.; Robbins, D.; Schmidt, C.; Burczak, J.; Croce, C.M.; Copeland, T.; et al. hMSH5: A human MutS homologue that forms a novel heterodimer with hMSH4 and is expressed during spermatogenesis. Cancer Res. 1999, 59, 816–822. [Google Scholar] [PubMed]
- Santucci-Darmanin, S.; Walpita, D.; Lespinasse, F.; Desnuelle, C.; Ashley, T.; Paquis-Flucklingeret, V. MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J. 2000, 14, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.; Cohen, P. Novel and diverse functions of the DNA mismatch repair family in mammalian meiosis and recombination. Cytogenet. Genome Res. 2004, 107, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Her, C. MutS Homologues hMSH4 and hMSH5: Diverse Functional Implications in Humans. Front. Biosci. 2007, 12, 905. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.; Wu, X.; Her, C. MutS Homologues hMSH4 and hMSH5: Genetic Variations, Functions, and Implications in Human Diseases. Curr. Genom. 2013, 14, 81–90. [Google Scholar] [CrossRef]
- Chu, Y.-L.; Wu, X.; Xu, Y.; Her, C. MutS homologue hMSH4: Interaction with eIF3f and a role in NHEJ-mediated DSB repair. Mol. Cancer 2013, 12, 51. [Google Scholar] [CrossRef]
- Edelmann, W.; Cohen, P.E.; Kneitz, B.; Winand, N.; Lia, M.; Heyer, J.; Kolodner, R.; Pollard, J.W.; Kucherlapati, R. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat. Genet. 1999, 21, 123–127. [Google Scholar] [CrossRef]
- Kelly, K.O.; Dernburg, A.F.; Stanfield, G.M.; Villeneuve, A.M. Caenorhabditis elegans msh-5 is required for both normal and radiation-induced meiotic crossing over but not for completion of meiosis. Genetics 2000, 156, 617–630. [Google Scholar]
- Xu, Y.; Wu, X.; Her, C. hMSH5 Facilitates the Repair of Camptothecin-induced Double-strand Breaks through an Interaction with FANCJ*. J. Biol. Chem. 2015, 290, 18545–18558. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Figueroa, A.; Fragaki, K.; Destroismaisons, L.; Lacas-Gervais, S.; Lespinasse, F.; Vandenbos, F.; Pradelli, L.A.; Ricci, J.-E.; Rötig, A.; et al. The human MSH5 (MutS Homolog 5) protein localizes to mitochondria and protects the mitochondrial genome from oxidative damage. Mitochondrion 2012, 12, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.A.; Sakoda, L.C.; Loomis, M.M.; Barnett, M.J.; Julianto, L.; Thornquist, M.D.; Neuhouser, M.L.; Weiss, N.S.; Goodman, G.E.; Chen, C. DNA repair genotype and lung cancer risk in the beta-carotene and retinol efficacy trial. Int. J. Mol. Epidemiol. Genet. 2013, 4, 11–34. [Google Scholar] [PubMed]
- Timofeeva, M.N.; Hung, R.J.; Rafnar, T.; Christiani, D.C.; Field, J.K.; Bickeböller, H.; Risch, A.; McKay, J.D.; Wang, Y.; Dai, J.; et al. Influence of common genetic variation on lung cancer risk: Meta-analysis of 14 900 cases and 29 485 controls. Hum. Mol. Genet. 2012, 21, 4980–4995. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Wu, X.; Lee, T.-H.; Doggett, N.A.; Her, C. Two variants of MutS homolog hMSH5: Prevalence in humans and effects on protein interaction. Biochem. Biophys. Res. Commun. 2005, 332, 524–532. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Qian, C.-Y.; Gao, Y.-F.; Chen, J.; Zhou, H.; Yin, J.-Y. Association between DNA mismatch repair gene polymorphisms and platinum-based chemotherapy toxicity in non-small cell lung cancer patients. Chin. J. Cancer 2017, 36, 12. [Google Scholar] [CrossRef]
- Kazma, R.; Babron, M.-C.; Gaborieau, V.; Génin, E.; Brennan, P.; Hung, R.J.; McLaughlin, J.R.; Krokan, H.E.; Elvestad, M.B.; Skorpen, F.; et al. Lung cancer and DNA repair genes: Multilevel association analysis from the International Lung Cancer Consortium. Carcinogenesis 2012, 33, 1059–1064. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Y.; Breitling, L.P.; Brenner, H. Tobacco smoking and methylation of genes related to lung cancer development. Oncotarget 2016, 7, 59017–59028. [Google Scholar] [CrossRef]
- Wang, Y.; Broderick, P.; Webb, E.L.; Wu, X.; Vijayakrishnan, J.; Matakidou, A.; Qureshi, M.; Dong, Q.; Gu, X.; Chen, W.V.; et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat. Genet. 2008, 40, 1407–1409. [Google Scholar] [CrossRef]
- Scarbrough, P.M.; Weber, R.P.; Iversen, E.S.; Brhane, Y.; Amos, C.I.; Kraft, P.; Hung, R.J.; Sellers, T.A.; Witte, J.S.; Pharoah, P.; et al. A Cross-Cancer Genetic Association Analysis of the DNA Repair and DNA Damage Signaling Pathways for Lung, Ovary, Prostate, Breast, and Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2015, 25, 193–200. [Google Scholar] [CrossRef]
- Blackwell, L.J.; Martik, D.; Bjornson, K.P.; Bjornson, E.S.; Modrich, P. Nucleotide-promoted Release of hMutSα from Heteroduplex DNA Is Consistent with an ATP-dependent Translocation Mechanism. J. Biol. Chem. 1998, 273, 32055–32062. [Google Scholar] [CrossRef] [PubMed]
- Kansikas, M.; Kariola, R.; Nystrom-Lahti, M. Verification of the three-step model in assessing the pathogenicity of mismatch repair gene variants. Hum. Mutat. 2010, 32, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Levine, E.A.; Franco, V.I.; Allen, G.O.; Gong, F.; Zhang, Y.; Hu, J.J. Combined Genetic and Nutritional Risk Models of Triple Negative Breast Cancer. Nutr. Cancer 2014, 66, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Zelga, P.; Przybyłowska-Sygut, K.; Zelga, M.; Dziki, A.; Majsterek, I. The 116G > A MSH6 and IVS1-1121C > T PMS2 Genes Polymorphisms Modulate the Risk of the Sporadic Colorectal Cancer Development in Polish Population. Pathol. Oncol. Res. 2017, 24, 231–235. [Google Scholar] [CrossRef]
- Zelga, P.; Przybylowska-Sygut, K.; Zelga, M.; Dziki, A.; Majsterek, I. Polymorphism of Gly39Glu (c.116G>A) hMSH6 is associated with sporadic colorectal cancer development in the Polish population: Preliminary results. Adv. Clin. Exp. Med. 2017, 26, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Silva, S.N.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; Rueff, J. Mismatch repair single nucleotide polymorphisms and thyroid cancer susceptibility. Oncol. Lett. 2018, 15, 6715–6726. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, M.; Wang, Y.; Veber, N.; Mwapagha, L.M.; Parker, M.I. The Cumulative Effects of Polymorphisms in the DNA Mismatch Repair Genes and Tobacco Smoking in Oesophageal Cancer Risk. PLoS ONE 2012, 7, e36962. [Google Scholar] [CrossRef]
- Curtin, K.; Samowitz, W.S.; Wolff, R.K.; Caan, B.J.; Ulrich, C.M.; Potter, J.D.; Slattery, M.L. MSH6 G39E polymorphism and CpG island methylator phenotype in colon cancer. Mol. Carcinog. 2009, 48, 989–994. [Google Scholar] [CrossRef]
- Dong, X.; Li, Y.; Chang, P.; Hess, K.R.; Abbruzzese, J.L.; Li, D. DNA mismatch repair network gene polymorphism as a susceptibility factor for pancreatic cancer. Mol. Carcinog. 2011, 51, 491–499. [Google Scholar] [CrossRef]
- Zanusso, C.; Bortolus, R.; Dreussi, E.; Polesel, J.; Montico, M.; Cecchin, E.; Gagno, S.; Rizzolio, F.; Arcicasa, M.; Novara, G.; et al. Impact of DNA repair gene polymorphisms on the risk of biochemical recurrence after radiotherapy and overall survival in prostate cancer. Oncotarget 2017, 8, 22863–22875. [Google Scholar] [CrossRef]
- Cecchin, E.; D’Andrea, M.; Lonardi, S.; Zanusso, C.; Pella, N.; Errante, D.; De Mattia, E.; Polesel, J.; Innocenti, F.; Toffoli, G. A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharm. J. 2012, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Doss, C.G.P.; Rao, S. Investigation on the role of nsSNPs in HNPCC genes—A bioinformatics approach. J. Biomed. Sci. 2009, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Hu, Z.; Shu, Y.; Pan, S.; Chen, W.; Wang, Y.; Hu, L.; Jiang, Y.; Dai, J.; Ma, H.; et al. Potentially functional polymorphisms in DNA repair genes and non-small-cell lung cancer survival: A pathway-based analysis. Mol. Carcinog. 2011, 51, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Lockman, J.C.; Frankel, W.L.; Hampel, H.; Steenblock, K.; Burgart, L.J.; Thibodeau, S.N.; De La Chapelle, A. Mismatch Repair GenePMS. Cancer Res. 2004, 64, 4721–4727. [Google Scholar] [CrossRef]
- Mann, A.; Høgdall, E.; Ramus, S.J.; DiCioccio, R.A.; Høgdall, C.; Quaye, L.; McGuire, V.; Whittemore, A.S.; Shah, M.; Greenberg, D.; et al. Mismatch repair gene polymorphisms and survival in invasive ovarian cancer patients. Eur. J. Cancer 2008, 44, 2259–2265. [Google Scholar] [CrossRef]
- Song, H.; Ramus, S.J.; Quaye, L.; DiCioccio, R.A.; Tyrer, J.; Lomas, E.; Shadforth, D.; Høgdall, E.V.S.; Høgdall, C.K.; McGuire, V.; et al. Common variants in mismatch repair genes and risk of invasive ovarian cancer. Carcinogenesis 2006, 27, 2235–2242. [Google Scholar] [CrossRef]
- Horii, A.; Han, H.; Sasaki, S.; Shimada, M.; Nakamura, Y. Cloning, Characterization and Chromosomal Assignment of the Human Genes Homologous to Yeast PMS1, a Member of Mismatch Repair Genes. Biochem. Biophys. Res. Commun. 1994, 204, 1257–1264. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Carter, K.C.; Shell, B.K.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W. Genomic Organization of the HumanPMS2Gene Family. Genomics 1995, 30, 195–206. [Google Scholar] [CrossRef]
- Kondo, E.; Horii, A.; Fukushige, S. The human PMS2L proteins do not interact with hMLH1, a major DNA mismatch repair protein. J. Biochem. 1999, 125, 818–825. [Google Scholar]
- De Vos, M.; Hayward, B.E.; Picton, S.; Sheridan, E.; Bonthron, D.T. Novel PMS2 Pseudogenes Can Conceal Recessive Mutations Causing a Distinctive Childhood Cancer Syndrome. Am. J. Hum. Genet. 2004, 74, 954–964. [Google Scholar] [CrossRef]
- Mitchell, R.J.; Farrington, S.M.; Dunlop, M.G.; Campbell, H. Mismatch Repair Genes hMLH1 and hMSH2 and Colorectal Cancer: A HuGE Review. Am. J. Epidemiol. 2002, 156, 885–902. [Google Scholar] [CrossRef] [PubMed]
- Kamory, E.; Kolacsek, O.; Ottó, S.; Csuka, O. hMLH1 and hMSH2 somatic inactivation mechanisms in sporadic colorectal cancer patients. Pathol. Oncol. Res. 2003, 9, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar] [PubMed]
- Geisler, J.P.; Goodheart, M.J.; Sood, A.K.; Holmes, R.J.; Hatterman-Zogg, M.A.; Buller, R.E. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer 2003, 98, 2199–2206. [Google Scholar] [CrossRef]
- Pierini, S.; Jordanov, S.H.; Mitkova, A.V.; Chalakov, I.J.; Melnicharov, M.B.; Kunev, K.V.; Mitev, V.I.; Kaneva, R.P.; Goranova, T.E. Promoter hypermethylation of CDKN2A, MGMT, MLH1, and DAPK genes in laryngeal squamous cell carcinoma and their associations with clinical profiles of the patients. Head Neck 2013, 36, 1103–1108. [Google Scholar] [CrossRef]
- Gomes, A.; Reis-Silva, M.; Alarcao, A.; Couceiro, P.; Sousa, V.; Carvalho, L. Promoter hypermethylation of DNA repair genes MLH1 and MSH2 in adenocarcinomas and squamous cell carcinomas of the lung. Rev. Por.T Pneumol. 2014, 20, 20–30. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Hampel, H.; Wu, C.; Weng, D.Y.; Shields, P.G.; Frankel, W.L.; Pan, X.; De La Chapelle, A.; Goldberg, R.; Bekaii-Saab, T. Patients with colorectal cancer associated with Lynch syndrome and MLH1 promoter hypermethylation have similar prognoses. Genet. Med. 2016, 18, 863–868. [Google Scholar] [CrossRef]
- Orimo, H.; Nakajima, E.; Yamamoto, M.; Ikejima, M.; Emi, M.; Shimada, T. Association between single nucleotide polymorphisms in the hMSH3 gene and sporadic colon cancer with microsatellite instability. J. Hum. Genet. 2000, 45, 228–230. [Google Scholar] [CrossRef]
- Gazzoli, I.; Kolodner, R.D. Regulation of the Human MSH6 Gene by the Sp1 Transcription Factor and Alteration of Promoter Activity and Expression by Polymorphisms. Mol. Cell. Biol. 2003, 23, 7992–8007. [Google Scholar] [CrossRef]
- Wu, S.; Chen, J.; Ji, Y.; Liu, Y.; Gao, L.; Chen, G.; Shen, K.; Huang, B. Association between the hMSH2 IVS12-6 T>C polymorphism and cancer risk: A meta-analysis. Exp. Ther. Med. 2011, 2, 1193–1198. [Google Scholar] [CrossRef]
- Yuan, Z.Q.; Gottlieb, B.; Beitel, L.K.; Wong, N.; Gordon, P.H.; Wang, Q.; Puisieux, A.; Foulkes, W.D.; Trifiro, M. Polymorphisms and HNPCC: PMS2-MLH1 protein interactions diminished by single nucleotide polymorphisms. Hum. Mutat. 2002, 19, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Turner, D.; Masciullo, V.; Schepis, F.; Albino, D.; Daniel, R.; Skalka, A.M.; Meropol, N.J.; Alberti, C.; LaRue, L.; et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc. Natl. Acad. Sci. USA 2003, 100, 15071–15076. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Khattar, N.H.; Gu, L.; Li, G.-M. Roles of mismatch repair proteins hMSH2 and hMLH1 in the development of sporadic breast cancer. Cancer Lett. 2005, 223, 143–150. [Google Scholar] [CrossRef]
- Russo, A.; Corsale, S.; Cammareri, P.; Agnese, V.; Cascio, S.; Di Fede, G.; Macaluso, M.; Bazan, V. Pharmacogenomics in colorectal carcinomas: Future perspectives in personalized therapy. J. Cell. Physiol. 2005, 204, 742–749. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P. DNA Mismatch Repair: Functions and Mechanisms. Chem. Rev. 2006, 37, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Worrillow, L.J.; Travis, L.B.; Smith, A.G.; Rollinson, S.; Smith, A.J.; Wild, C.P.; Holowaty, E.J.; Kohler, B.A.; Wiklund, T.; Pukkala, E.; et al. An intron splice acceptor polymorphism in hMSH2 and risk of leukemia after treatment with chemotherapeutic alkylating agents. Clin. Cancer Res. 2003, 9, 3012–3020. [Google Scholar] [PubMed]
- Park, J.H.; Kim, N.S.; Park, J.Y.; Chae, Y.S.; Kim, J.G.; Sohn, S.K.; Moon, J.-H.; Kang, B.W.; Ryoo, H.M.; Bae, S.H.; et al. MGMT −535G>T polymorphism is associated with prognosis for patients with metastatic colorectal cancer treated with oxaliplatin-based chemotherapy. J. Cancer Res. Clin. Oncol. 2010, 136, 1135–1142. [Google Scholar] [CrossRef]
- Kim, J.G.; Chae, Y.S.; Sohn, S.K.; Moon, J.H.; Kang, B.W.; Park, J.Y.; Jeon, S.W.; Lee, M.-H.; Lim, K.-H.; Choi, G.S.; et al. IVS10+12A>G polymorphism in hMSH2 gene associated with prognosis for patients with colorectal cancer. Ann. Oncol. 2010, 21, 525–529. [Google Scholar] [CrossRef]
- Jung, C.Y.; Choi, J.E.; Park, J.M.; Chae, M.H.; Kang, H.-G.; Kim, K.M.; Lee, S.J.; Lee, W.-K.; Kam, S.; Cha, S.I.; et al. Polymorphisms in the hMSH2 Gene and the Risk of Primary Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 762–768. [Google Scholar] [CrossRef]
- Boeckmann, L.; Thoms, K.-M.; Gutzmer, R.; Has, C.; Kunz, M.; Kuschal, C.; Laspe, P.; Struever, D.; Emmert, S. Modulation of the efficacy of temozolomide and dacarbazine melanoma treatment by DNA-repair factors in vivo and in vitro. Int. J. Clin. Pharmacol. Ther. 2009, 47, 33–35. [Google Scholar] [CrossRef]
- Vymetalkova, V.; Slyskova, J.; Korenková, V.; Bielik, L.; Langerová, L.; Procházka, P.; Rejhova, A.; Schwarzová, L.; Pardini, B.; Naccarati, A.; et al. Molecular characteristics of mismatch repair genes in sporadic colorectal tumors in Czech patients. BMC Med. Genet. 2014, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Tomasova, K.; Cumova, A.; Seborova, K.; Horak, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713.