The tumour microenvironment (TME) comprises a complex ecosystem of different cell types, including immune cells, cells of the vasculature and lymphatic system, cancer-associated fibroblasts, pericytes, and adipocytes. Cancer proliferation, invasion, metastasis, drug resistance and immune escape are all influenced by the dynamic interaction between cancer cells and TME. Microbes, such as bacteria, fungi, viruses, archaea and protists, found within tumour tissues, constitute the intratumour microbiota, which is tumour type-specific and distinct among patients with different clinical outcomes. Growing evidence reveals a significant relevance of local microbiota in the colon, liver, breast, lung, oral cavity and pancreas carcinogenesis. Moreover, there is a growing interest in the tumour immune microenvironment (TIME) pointed out in several cross-sectional studies on the correlation between microbiota and TME. It is now known that microorganisms have the capacity to change the density and function of anticancer and suppressive immune cells, enabling the promotion of an inflammatory environment. As immunotherapy (such as immune checkpoint inhibitors) is becoming a promising therapy using TIME as a therapeutic target, the analysis and comprehension of local microbiota and its modulating strategies can help improve cancer treatments.
- microbiota
- microbiome
- cancer
- breast cancer
- immune system
- immune microenvironment
- treatment
- immunotherapy
1. Introduction
2. Human Microbiota and the Relation with the Host
Near 100 trillion dynamic microorganisms from 5000 different species, including bacteria, viruses, fungi, archaea and protists, inhabit the human body in different locations, such as the gastrointestinal tract, skin, vaginal mucosa or the oral cavity and play different roles in immune system regulation, inflammatory state, tolerance for commensal bacteria, recognition of potentially infectious pathogenic organisms, intestinal permeability, energy balance and endocrine hormone secretion. The “microbiota” refers to the set composed of resident microbes on and inside the body, and the “microbiome” is the collective genome of these biological agents [1][8][1,8]. Human microbiota composition is distinctive to each individual, probably starting before birth. There is growing evidence that placenta, amniotic fluid and meconium microbial flora include non-pathogenic commensal microbes, which probably contributes to a possible heritage of maternal microbiota and foetal immune system development [9][10][11][9,10,11]. Acquisition of significant amounts of microbiota occurs during and immediately after birth and develops during the first three or four years of life, influenced by breastfed, household exposures, chronic conditions and geographic location. After that period, microbiota composition becomes relatively stable, only slightly modified throughout adulthood by host genetics, diet, lifestyle and diseases [9][10][9,10]. Regarding microbiota, its complexity can be described using the concepts of alpha-diversity, that describes the richness in a given sample (i.e., number of organisms and distribution of those organisms), and beta-diversity, that defines the extent of relative or absolute overlap of a microbial community between different samples [12]. Resilience is related to microbiota capacity for self-regeneration and restoration of homeostasis after any shift in its composition. However, in some cases, the microbiota cannot remain resilient after a perturbation, leading to a new equilibrium state, called “dysbiosis”. Dysbiosis, an altered composition of commensal microbiota and its metabolic activity, causes an imbalance in the symbiosis between the host and its organic habitat. Therefore, this deregulation can harm the human host and influence the onset of various inflammatory, auto-immune or malignant conditions [4][9][4,9]. For that reason, human resident microbiota and its complex relation with the host are now emerging as important elements in the lifelong maintenance of health and immune system homeostasis, with substantial attention given to its influence on cancer cell proliferation, tumourigenesis, disease progression and treatment outcomes [3][4][3,4]. Despite centuries of historical reports linking cancer and microbes, the International Agency for Research on Cancer (IACR) just considers 11 of the ~1012 microbial species on earth to directly cause cancer. However, it is suggested that approximately 20% of human cancers may be linked to microbial pathogens [12]. Several oncogenic microbes drive cancer, with Helicobacter-pylori-induced gastritis and gastric adenocarcinoma being perhaps the best evidence that the microbiota is not just a bystander in the cancer development process [13]. H. pylori. infection can contribute to the release of virulence factors that cause cellular stress in gastric epithelium, affecting host cell signalling pathways. Eradication of H. pylori. is an important method of reducing the risk of gastric cancer [14]. Previous studies have also established a causal link between the gut bacterium Bacteroides fragilis, oral pathogen Fusobacterium nucleatum, Escherichia coli and colorectal cancer [15][16][17][18][15,16,17,18]. The presence of F. nucleatum is associated with malignant transformation of colorectal adenomas to carcinoma and is also related to a worse survival of colorectal patients [14]. Metagenomic sequencing studies have detected significant differences in the composition of microbial communities in numerous human cancers compared to controls with normal tissues [12][19][20][12,19,20]. Although not fully clarified, various mechanisms of dysbiosis-induced cancer have been proposed in several studies: induction of inflammatory microenvironment and epithelial–mesenchymal transition (EMT), increase in reactive oxygen species (ROS) and DNA damage, genotoxic substances gathering, suppression of antitumour immune response and destruction of the gut mucosal layer with changes in intestinal permeability that allows translocation of pathogens and its byproducts to surrounding tissues and systemic circulation [1][21][22][23][24][25][1,22,23,24,25,26]. Although microbiota influences carcinogenesis through mechanisms independent of inflammation and immune system, the most recognised link is between microbiota and cancer via its effects on innate and adaptive immunity, modulating both local and systemic immune responses of the host [1][2][26][1,2,27]. This association is particularly strong between the gut microbiota and intestinal mucosal immune system. Pattern recognition receptors (PRRs), like Toll-like receptors (TLRs), are expressed in the human body by many cells, including immune cells, and act as detectors of pathogen components. Through microbe- or pathogen-associated molecular patterns (MAMPs or PAMPs), microbes interact with these receptors, activating inflammatory pathways and causing a cytokine release [1][2][26][1,2,27]. In addition, bacterial metabolites and byproducts also directly interfere with immune local cells’ actions, stimulating the maturation of local dendritic cells (DCs) through interaction with PRRs. These cells travel from their area to mesenteric lymph nodes, triggering lymphocyte differentiation of naïve CD4+ T cells into regulatory T-lymphocytes (Tregs) and T helper 17 (Th17). After maturation, effectors T cells can travel back to their original place to regulate local immune responses while another subset migrates to systemic circulation and influences immunity in different sites. For example, circulating Th17 cells enhance antitumour immunity, protecting against bacterial and fungal infections and circulating Tregs secrete anti-inflammatory cytokines. Another example of a direct link between microbiota and local immune response is its impact on B cells as the main mediator of gut mucosal homeostasis through the production of immunoglobulin A, which blocks bacterial adherence to epithelial cells [1][2][26][1,2,27]. Environmental factors such as inappropriate diet patterns are also important contributors to alterations in microbiota diversity. Microbes use ingested nutrients for harvesting energy and basic biological processes. Consumption of high levels of red meat is a risk factor for colorectal cancer and several other cancers by various mechanisms, some of them dependent on gut bacteria. Increased colonic protein levels intake can lead to increased bacterial fermentation of amino acids to N-nitroso compounds that induce DNA alkylation and mutations in the host. High fibre, low-fat diets are also capable of shifting the microbiota community towards the advantageous bacteria and increasing microbiota-derived short-chain fatty acids (SCFAs), like butyrate, a pleiotropic molecule that exerts its tumour-suppressive properties by multiple mechanisms and has been implicated in colorectal cancer prevention based on metagenomic studies and mouse models [1][12][1,12]. Faecal microbiota transplant (FMT) is an emerging therapeutic approach in many potential applications and has primarily been applied in patients with relapsed/refractory Clostridioides difficile infection. Due to the complexity of the diseases and their treatment, patients with haematologic and oncologic diseases are particularly susceptible to complications related to altered intestinal microbiota [27][28]. Currently, there are nearly 40 studies registered that primarily evaluate the safety of FMT, the use of FMT following allogeneic hematopoietic stem cell transplantation, improvement in ICI response, and the treatment of the complications that arise due to cancer therapy [27][28]. Various retrospective studies suggested a possible relation between broad-spectrum antibiotics, altered intestinal microbiota and its negative impact on responses to ICI treatment in cancer patients [28][29][30][31][29,30,31,32]. Based on these findings, two studies were performed, aiming to determine the safety and feasibility of FMT before re-introducing immunotherapy in refractory malignant melanoma. This treatment increased the intratumour immune activity in some patients, translated into objective clinical responses. These results support the concept of overcoming resistance to immunotherapy by modulating gut microbiota [32][33][33,34].3. Tumour Microenvironment
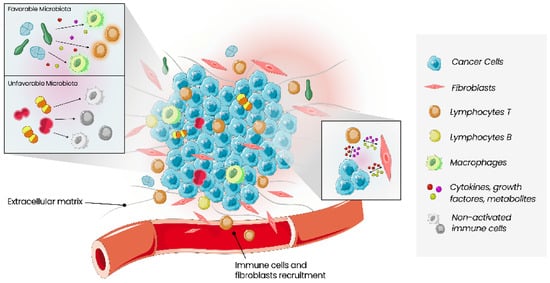
The tumour microenvironment (TME) relates to cancer cells and all types of cells surrounding them, including immune cells, blood vessels, extracellular matrix, fibroblasts, lymphocytes, signalling molecules such as cytokines, growth factors and enzymes (Figure 1) [34][35][35,36]. Interactions between these two types of cells, malignant and non-malignant, affect the tumour, the process of carcinogenesis, proliferation of malignant cells, and progression of the tumour. These interactions contribute to the host’s tolerance and response to the tumour. The mechanisms that allow tumour proliferation include angiogenesis, inhibition of apoptosis, immune system suppression, and are all controlled by cells of TME [5]. Growing evidence of this relationship between cancer and TME also increases the interest in TME as a prognostic factor and a potential therapeutic target [35][36].