Staphylococcus aureus (S. aureus) is part of the normal skin and nasal microbiota, with approximately 30% of the healthy adult population colonized mainly in the nasopharyngeal cavity. While colonization is usually asymptomatic, a symptomatic infection can occur if there is a breach in the mucosal barrier or skin. The severity of symptomatic infections ranges from superficial skin and soft tissue infections, to devastating complications, such as necrotizing pneumonia, endocarditis, toxic shock syndrome, and sepsis. In the pre-antibiotic era, S. aureus bacteremia mortality rates were astonishingly high, ranging between 75% and 83%. Even though antibiotics have reduced this number significantly, S. aureus bloodstream infections still account for over 19,000 deaths annually in the United States. With every new antibiotic that is developed, S. aureus resistance has been quickly observed. MRSA strains that are resistant to all penicillin-like β-lactam antibiotics pose a particularly serious threat to the community. Two types of MRSA exist: hospital-acquired (HA-)MRSA and community-acquired (CA-)MRSA. CA-MRSA strains are typically regarded as more virulent and can cause infections in otherwise healthy individuals. This notion is further supported by experimental animal studies, whereas HA-MRSA strains are less virulent than CA-MRSA and cause fewer disseminating diseases. Although in animal models, mice are typically not treated with antibiotics, which is unlike a hospital setting.
- Staphylococcus aureus
- MRSA
- antimicrobial therapy
1. The Infectious Cycle of S. aureus in Non-Professional Phagocytes
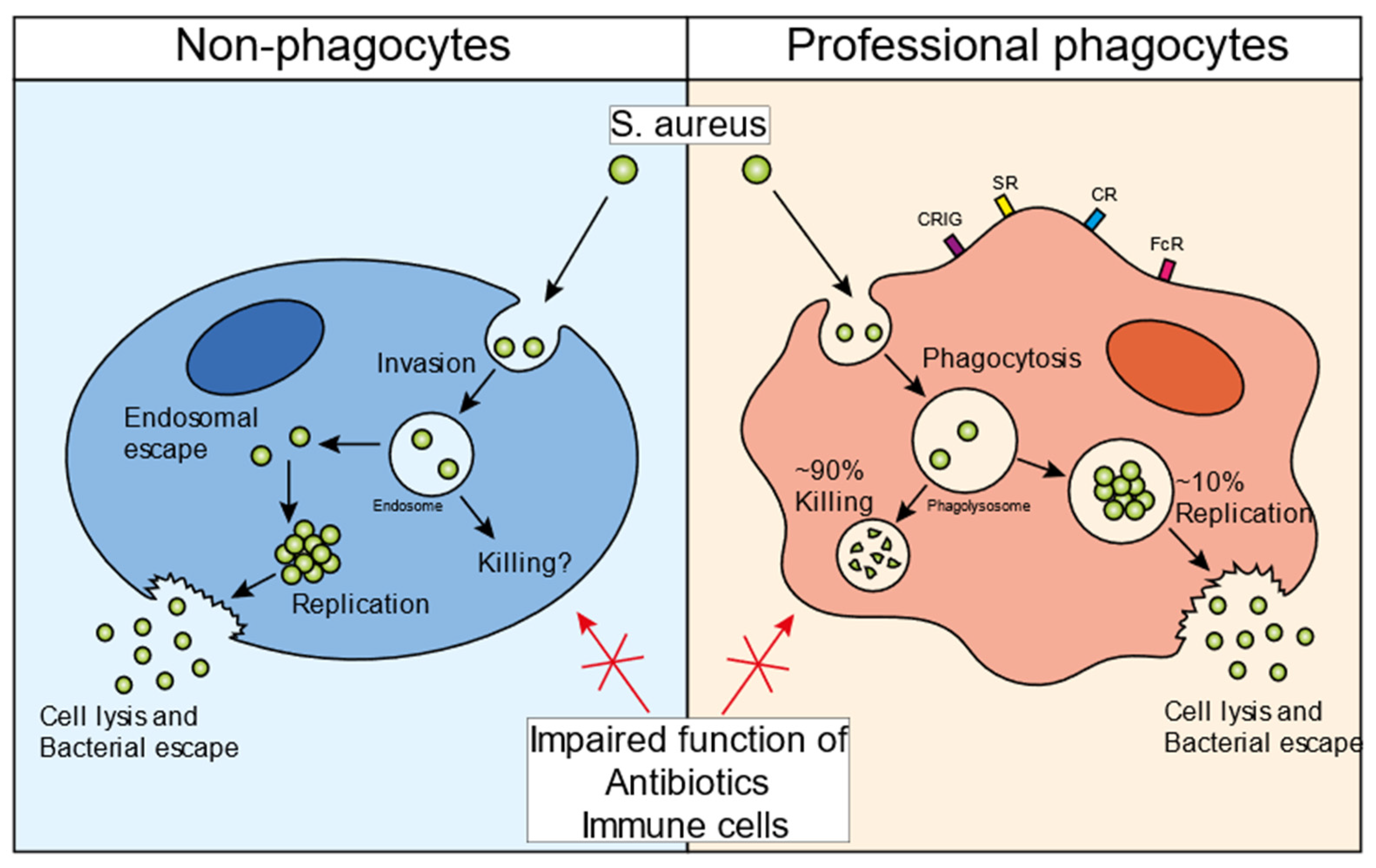
2. The Infectious Cycle of S. aureus in Professional Phagocytes
3. Intraphagolysosomal Evasion Strategies of S. aureus
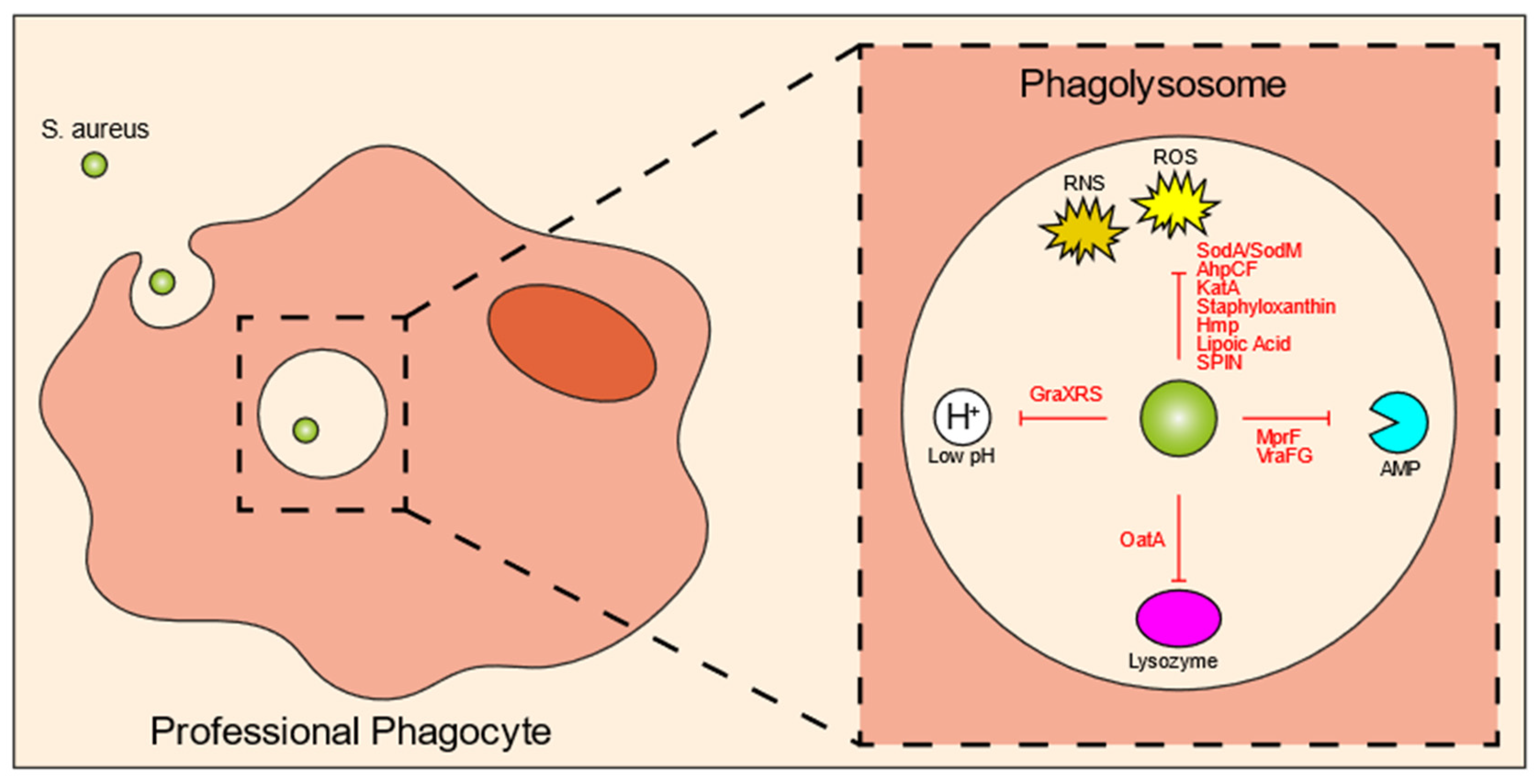
34. Bacterial Specificity of the Intracellular Reservoir
References
- Bayles, K.W.; Wesson, C.A.; Liou, L.E.; Fox, L.K.; Bohach, G.A.; Trumble, W.R. Intracellular Staphylococcus aureus Escapes the Endosome and Induces Apoptosis in Epithelial Cells. Infect. Immun. 1998, 66, 336–342.
- Hess, D.J.; Henry-Stanley, M.J.; Erickson, E.A.; Wells, C.L. Intracellular Survival of Staphylococcus aureus within Cultured Enterocytes. J. Surg. Res. 2003, 114, 42–49.
- Menzies, B.E.; Kourteva, I. Staphylococcus aureus α-Toxin Induces Apoptosis in Endothelial Cells. FEMS Immunol. Med. Microbiol. 2000, 29, 39–45.
- Nair, S.P.; Bischoff, M.; Senn, M.M.; Berger-Bächi, B. The ΣB Regulon Influences Internalization of Staphylococcus aureus by Osteoblasts. Infect. Immun. 2003, 71, 4167–4170.
- Rogers, R.; Tompsett, R. The Survival of Staphylococci within Human Leukocytes. J. Exp. Med. 1952, 28, 470.
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, Invasion and Evasion: The Many Functions of the Surface Proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62.
- Moldovan, A.; Fraunholz, M.J. In or out: Phagosomal Escape of Staphylococcus aureus. Cell. Microbiol. 2019, 21, e12997.
- Qazi, S.N.A.; Harrison, S.E.; Self, T.; Williams, P.; Hill, P.J. Real-Time Monitoring of Intracellular Staphylococcus aureus Replication. J. Bacteriol. 2004, 186, 1065–1077.
- Shompole, S.; Henon, K.T.; Liou, L.E.; Dziewanowska, K.; Bohach, G.A.; Bayles, K.W. Biphasic Intracellular Expression of Staphylococcus aureus Virulence Factors and Evidence for Agr-Mediated Diffusion Sensing. Mol. Microbiol. 2003, 49, 919–927.
- Schnaith, A.; Kashkar, H.; Leggio, S.A.; Addicks, K.; Krönke, M.; Krut, O. Staphylococcus aureus Subvert Autophagy for Induction of Caspase-Independent Host Cell Death. J. Biol. Chem. 2007, 282, 2695–2706.
- Queck, S.Y.; Jameson-Lee, M.; Villaruz, A.E.; Bach, T.H.L.; Khan, B.A.; Sturdevant, D.E.; Ricklefs, S.M.; Li, M.; Otto, M. RNAIII-Independent Target Gene Control by the Agr Quorum-Sensing System: Insight into the Evolution of Virulence Regulation in Staphylococcus aureus. Mol. Cell 2008, 32, 150–158.
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.C.; Schäfer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; et al. Cytoplasmic Replication of Staphylococcus aureus upon Phagosomal Escape Triggered by Phenol-Soluble Modulin α. Cell. Microbiol. 2014, 16, 451–465.
- Surewaard, B.G.J.; Nijland, R.; Spaan, A.N.; Kruijtzer, J.A.W.; de Haas, C.J.C.; van Strijp, J.A.G. Inactivation of Staphylococcal Phenol Soluble Modulins by Serum Lipoprotein Particles. PLoS Pathog. 2012, 8, e1002606.
- Hommes, J.W.; Kratofil, R.M.; Wahlen, S.; de Haas, C.J.C.; Hildebrand, R.B.; Hovingh, G.K.; Otto, M.; van Eck, M.; Hoekstra, M.; Korporaal, S.J.A.; et al. High Density Lipoproteins Mediate in Vivo Protection against Staphylococcal Phenol-Soluble Modulins. Sci. Rep. 2021, 11, 15357.
- Gresham, H.D.; Lowrance, J.H.; Caver, T.E.; Wilson, B.S.; Cheung, A.L.; Lindberg, F.P. Survival of Staphylococcus aureus Inside Neutrophils Contributes to Infection. J. Immunol. 2000, 164, 3713–3722.
- Koziel, J.; Maciag-Gudowska, A.; Mikolajczyk, T.; Bzowska, M.; Sturdevant, D.E.; Whitney, A.R.; Shaw, L.N.; DeLeo, F.R.; Potempa, J. Phagocytosis of Staphylococcus aureus by Macrophages Exerts Cytoprotective Effects Manifested by the Upregulation of Antiapoptotic Factors. PLoS ONE 2009, 4, e5210.
- Spaan, A.N.; Surewaard, B.G.J.; Nijland, R.; van Strijp, J.A.G. Neutrophils Versus Staphylococcus aureus: A Biological Tug of War. Annu. Rev. Microbiol. 2013, 67, 629–650.
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and Tissue Specialization. Annu. Rev. Immunol. 2015, 33, 643–675; 0324141122.
- Rogers, D.E. Studies on Bacteriemia I. Mechanisms Relating to the Persistence of Bacteriemia in Rabbits Following the Intravenous Injection of Staphylococci. J. Exp. Med. 1956, 103, 713–742.
- Thwaites, G.E.; Gant, V. Are Bloodstream Leukocytes Trojan Horses for the Metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 2011, 9, 215–222.
- Krezalek, M.A.; Hyoju, S.; Zaborin, A.; Okafor, E.; Chandrasekar, L.; Bindokas, V.; Guyton, K.; Montgomery, C.P.; Daum, R.S.; Zaborina, O.; et al. Can Methicillin-Resistant Staphylococcus aureus Silently Travel from the Gut to the Wound and Cause Postoperative Infection? Modeling the “Trojan Horse Hypothesis”. Ann. Surg. 2018, 267, 749–758.
- Venditti, M.; Falcone, M.; Micozzi, A.; Carfagna, P.; Taglietti, F.; Serra, P.F.; Martino, P. Staphylococcus aureus Bacteremia in Patients with Hematologic Malignancies: A Retrospective Case-Control Study. Haematologica 2003, 88, 923–930.
- Krenkel, O.; Tacke, F. Liver Macrophages in Tissue Homeostasis and Disease. Nat. Rev. Immunol. 2017, 17, 306–321.
- Jenne, C.N.; Kubes, P. Immune Surveillance by the Liver. Nat. Immunol. 2013, 14, 996–1006.
- Pollitt, E.J.G.; Szkuta, P.T.; Burns, N.; Foster, S.J. Staphylococcus aureus Infection Dynamics. PLoS Pathog. 2018, 14, e1007112.
- Surewaard, B.G.J.; Deniset, J.F.; Zemp, F.J.; Amrein, M.; Otto, M.; Conly, J.; Omri, A.; Yates, R.M.; Kubes, P. Identification and Treatment of the Staphylococcus aureus Reservoir in Vivo. J. Exp. Med. 2016, 213, 1141–1151.
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens 2015, 4, 826–868.
- Lacoma, A.; Cano, V.; Moranta, D.; Regueiro, V.; Domínguez-Villanueva, D.; Laabei, M.; González-Nicolau, M.; Ausina, V.; Prat, C.; Bengoechea, J.A. Investigating Intracellular Persistence of Staphylococcus aureus within a Murine Alveolar Macrophage Cell Line. Virulence 2017, 8, 1761–1775.
- Tranchemontagne, Z.R.; Camire, R.B.; O’Donnell, V.J.; Baugh, J.; Burkholder, K.M. Staphylococcus aureus Strain USA300 Perturbs Acquisition of Lysosomal Enzymes and Requires Phagosomal Acidification for Survival inside Macrophages. Infect. Immun. 2015, 84, 241–253.
- Jubrail, J.; Morris, P.; Bewley, M.A.; Stoneham, S.; Johnston, S.A.; Foster, S.J.; Peden, A.A.; Read, R.C.; Marriott, H.M.; Dockrell, D.H. Inability to Sustain Intraphagolysosomal Killing of Staphylococcus aureus Predisposes to Bacterial Persistence in Macrophages. Cell. Microbiol. 2016, 18, 80–96.
- Surewaard, B.G.J.; Thanabalasuriar, A.; Zeng, Z.; Tkaczyk, C.; Cohen, T.S.; Bardoel, B.W.; Jorch, S.K.; Deppermann, C.; Bubeck Wardenburg, J.; Davis, R.P.; et al. α-Toxin Induces Platelet Aggregation and Liver Injury during Staphylococcus aureus Sepsis. Cell Host Microbe 2018, 24, 271–284.
- Jorch, S.K.; Surewaard, B.G.J.; Hossain, M.; Peiseler, M.; Deppermann, C.; Deng, J.; Bogoslowski, A.; van der Wal, F.; Omri, A.; Hickey, M.J.; et al. Peritoneal GATA6+ Macrophages Function as a Portal for Staphylococcus aureus Dissemination. J. Clin. Investig. 2019, 129, 4643–4656.
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-Induced Necroptosis Is a Major Mechanism of Staphylococcus aureus Lung Damage. PLoS Pathog. 2015, 11, e1004820.
- Flannagan, R.S.; Cosío, G.; Grinstein, S. Antimicrobial Mechanisms of Phagocytes and Bacterial Evasion Strategies. Nat. Rev. Microbiol. 2009, 7, 355–366.
- Kahl, B.C.; Goulian, M.; Van Wamel, W.; Herrmann, M.; Simon, S.M.; Kaplan, G.; Peters, G.; Cheung, A.L. Staphylococcus aureus RN6390 Replicates and Induces Apoptosis in a Pulmonary Epithelial Cell Line. Infect. Immun. 2000, 68, 5385–5392.
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532.
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Intracellular Replication of Staphylococcus aureus in Mature Phagolysosomes in Macrophages Precedes Host Cell Death, and Bacterial Escape and Dissemination. Cell. Microbiol. 2016, 18, 514–535.
- Jarry, T.M.; Cheung, A.L. Staphylococcus aureus Escapes More Efficiently from the Phagosome of a Cystic Fibrosis Bronchial Epithelial Cell Line than from Its Normal Counterpart. Infect. Immun. 2006, 74, 2568–2577.
- Lâm, T.T.; Giese, B.; Chikkaballi, D.; Kühn, A.; Wolber, W.; Pané-Farré, J.; Schäfer, D.; Engelmann, S.; Fraunholz, M.; Sinha, B. Phagolysosomal Integrity Is Generally Maintained after Staphylococcus aureus Invasion of Nonprofessional Phagocytes but Is Modulated by Strain 6850. Infect. Immun. 2010, 78, 3392–3393.
- Karavolos, M.H.; Horsburgh, M.; Ingham, E.; Foster, S.J. Role and Regulation of the Superoxide Dismutases of Staphylococcus aureus. Microbiology 2003, 149, 2749–2758.
- Das, D.; Saha, S.S.; Bishayi, B. Intracellular Survival of Staphylococcus aureus: Correlating Production of Catalase and Superoxide Dismutase with Levels of Inflammatory Cytokines. Inflamm. Res. 2008, 57, 340–349.
- Cosgrove, K.; Coutts, G.; Jonsson, I.M.; Tarkowski, A.; Kokai-Kun, J.F.; Mond, J.J.; Foster, S.J. Catalase (KatA) and Alkyl Hydroperoxide Reductase (AhpC) Have Compensatory Roles in Peroxide Stress Resistance and Are Required for Survival, Persistence, and Nasal Colonization in Staphylococcus aureus. J. Bacteriol. 2007, 189, 1025–1035.
- Mashruwala, A.A.; Boyd, J.M. The Staphylococcus aureus SrrAB Regulatory System Modulates Hydrogen Peroxide Resistance Factors, Which Imparts Protection to Aconitase during Aerobic Growth. PLoS ONE 2017, 12, e0170283.
- Pandey, S.; Sahukhal, G.S.; Elasri, M.O. The MsaABCR Operon Regulates the Response to Oxidative Stress in Staphylococcus aureus. J. Bacteriol. 2019, 201, e00417-19.
- Nobre, L.S.; Gonçalves, V.L.; Saraiva, L.M. Flavohemoglobin of Staphylococcus aureus. Methods Enzymol. 2008, 436, 203–216.
- Grayczyk, J.P.; Alonzo, F., III. Staphylococcus aureus Lipoic Acid Synthesis Limits Macrophage Reactive Oxygen and Nitrogen Species Production to Promote Survival during Infection. Infect. Immun. 2019, 87, e00344-19.
- De Jong, N.W.M.; Ramyar, K.X.; Guerra, F.E.; Nijland, R.; Fevre, C.; Voyich, J.M.; McCarthy, A.J.; Garcia, B.L.; Van Kessel, K.P.M.; Van Strijp, J.A.G.; et al. Immune Evasion by a Staphylococcal Inhibitor of Myeloperoxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 9439–9444.
- Leliefeld, P.H.C.; Pillay, J.; Vrisekoop, N.; Heeres, M.; Tak, T.; Kox, M.; Rooijakkers, S.H.M.; Kuijpers, T.W.; Pickkers, P.; Leenen, L.P.H.; et al. Differential Antibacterial Control by Neutrophil Subsets. Blood Adv. 2018, 2, 1344–1354.
- Villanueva, M.; García, B.; Valle, J.; Rapún, B.; Ruiz De Los Mozos, I.; Solano, C.; Martí, M.; Penadés, J.R.; Toledo-Arana, A.; Lasa, I. Sensory Deprivation in Staphylococcus aureus. Nat. Commun. 2018, 9, 523.
- Flannagan, R.S.; Kuiack, R.C.; McGavin, M.J.; Heinrichs, D.E. Staphylococcus aureus Uses the GraXRS Regulatory System To Sense and Adapt to the Acidified Phagolysosome in Macrophages. MBio 2018, 9, e01143-18.
- Chan, P.F.; Foster, S.J.; Ingham, E.; Clements, M.O. The Staphylococcus aureus Alternative Sigma Factor σ(B) Controls the Environmental Stress Response but Not Starvation Survival or Pathogenicity in a Mouse Abscess Model. J. Bacteriol. 1998, 180, 6082–6089.
- Olivier, A.C.; Lemaire, S.; Van Bambeke, F.; Tulkens, P.M.; Oldfield, E. Role of RsbU and Staphyloxanthin in Phagocytosis and Intracellular Growth of Staphylococcus aureus in Human Macrophages and Endothelial Cells. J. Infect. Dis. 2009, 200, 1367–1370.
- Bera, A.; Biswas, R.; Herbert, S.; Götz, F. The Presence of Peptidoglycan O-Acetyltransferase in Various Staphylococcal Species Correlates with Lysozyme Resistance and Pathogenicity. Infect. Immun. 2006, 74, 4598–4604.
- Shimada, T.; Park, B.G.; Wolf, A.J.; Brikos, C.; Goodridge, H.S.; Becker, C.A.; Reyes, C.N.; Miao, E.A.; Aderem, A.; Götz, F.; et al. Staphylococcus aureus Evades Lysozyme-Based Peptidoglycan Digestion That Links Phagocytosis, Inflammasome Activation, and IL-1β Secretion. Cell Host Microbe 2010, 7, 38–49.
- Kristian, S.A.; Dürr, M.; Van Strijp, J.A.G.; Neumeister, B.; Peschel, A. MprF-Mediated Lysinylation of Phospholipids in Staphylococcus aureus Leads to Protection against Oxygen-Independent Neutrophil Killing. Infect. Immun. 2003, 71, 546–549.
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; et al. Staphylococcus aureus Resistance to Human Defensins and Evasion of Neutrophil Killing via the Novel Virulence Factor MprF Is Based on Modification of Membrane Lipids with L-Lysine. J. Exp. Med. 2001, 193, 1067–1076.
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Götz, F. Inactivation of the Dlt Operon in Staphylococcus aureus Confers Sensitivity to Defensins, Protegrins, and Other Antimicrobial Peptides. J. Biol. Chem. 1999, 274, 8405–8410.
- Li, M.; Cha, D.J.; Lai, Y.; Villaruz, A.E.; Sturdevant, D.E.; Otto, M. The Antimicrobial Peptide-Sensing System Aps of Staphylococcus aureus. Mol. Microbiol. 2007, 66, 1136–1147.
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; et al. A Potential New Pathway for Staphylococcus aureus Dissemination: The Silent Survival of S. Aureus Phagocytosed by Human Monocyte-Derived Macrophages. PLoS ONE 2008, 3, e1409.
- Surewaard, B.; de Haas, C.; Vervoort, F.; Rigby, K.; DeLeo, F.; Otto, M.; van Strijp, J.; Nijland, R. Staphylococcal Alpha-Phenol Soluble Modulins Contribute to Neutrophil Lysis after Phagocytosis. Cell Microbiol. 2013, 15, 1427–1437.
- Geiger, T.; Francois, P.; Liebeke, M.; Fraunholz, M.; Goerke, C.; Krismer, B.; Schrenzel, J.; Lalk, M.; Wolz, C. The Stringent Response of Staphylococcus aureus and Its Impact on Survival after Phagocytosis through the Induction of Intracellular PSMs Expression. PLoS Pathog. 2012, 8, e1003016.
- Flannagan, R.S.; Watson, D.W.; Surewaard, B.G.J.; Kubes, P.; Heinrichs, D.E. The Surreptitious Survival of the Emerging Pathogen Staphylococcus Lugdunensis within Macrophages as an Immune Evasion Strategy. Cell. Microbiol. 2018, 20, e12869.
- Boldock, E.; Surewaard, B.G.J.; Shamarina, D.; Na, M.; Fei, Y.; Ali, A.; Williams, A.; Pollitt, E.J.G.; Szkuta, P.; Morris, P.; et al. Human Skin Commensals Augment Staphylococcus aureus Pathogenesis. Nat. Microbiol. 2018, 3, 881–890.
- Gibson, J.F.; Pidwill, G.R.; Carnell, O.T.; Surewaard, B.G.J.; Shamarina, D.; Sutton, J.A.F.; Jeffery, C.; Derré-Bobillot, A.; Archambaud, C.; Siggins, M.K.; et al. Commensal Bacteria Augment Staphylococcus aureus Infection by Inactivation of Phagocyte-Derived Reactive Oxygen Species. PLoS Pathog. 2021, 17, e1009880.
- Davis, J.M.; Ramakrishnan, L. The Role of the Granuloma in Expansion and Dissemination of Early Tuberculous Infection. Cell 2009, 136, 37–49.
- Drevets, D.A. Dissemination of Listeria Monocytogenes by Infected Phagocytes. Infect. Immun. 1999, 67, 3512–3517.
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In Vivo Imaging Reveals an Essential Role for Neutrophils in Leishmaniasis Transmitted by Sand Flies. Science 2008, 321, 970–975.
- Shi, M.; Li, S.S.; Zheng, C.; Jones, G.J.; Kim, K.S.; Zhou, H.; Kubes, P.; Mody, C.H. Real-Time Imaging of Trapping and Urease-Dependent Transmigration of Cryptococcus Neoformans in Mouse Brain. J. Clin. Investig. 2010, 120, 1683–1693.
- Sedlyarov, V.; Eichner, R.; Girardi, E.; Essletzbichler, P.; Goldmann, U.; Nunes-Hasler, P.; Srndic, I.; Moskovskich, A.; Heinz, L.X.; Kartnig, F.; et al. The Bicarbonate Transporter SLC4A7 Plays a Key Role in Macrophage Phagosome Acidification. Cell Host Microbe 2018, 23, 766–774.
- Xiong, Y.Q.; Fowler, V.G.; Yeaman, M.R.; Perdreau-Remington, F.; Kreiswirth, B.N.; Bayer, A.S. Phenotypic and Genotypic Characteristics of Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia In Vitro and in an Experimental Endocarditis Model. J. Infect. Dis. 2009, 199, 201–208.