Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Ernesto Solorzano.
Complex Lymphatic Anomalies (CLA) are characterized by idiopathic boney lesions caused by abnormal lymphatic invasion. The lymphatic network is a one-way system mainly responsible for maintaining fluid homeostasis, lipid absorption, and immune surveillance. CLAs include four diseases with overlapping and distinct clinical features: Gorham-Stout Disease (GSD), General Lymphatic Anomaly (GLA), Kaposiform Lymphangiomatosis (KLA), and Central Conducting Lymphatic Anomaly (CCLA).In order to highlight the known bone involvement of each CLA, their osteopathic phenotypes are described and illustrated below.
- lymphatic
- bone
- lymphangiogenesis
1. Gorham Stout Disease (GSD)
GSD, also known as “vanishing bone disease”, is an extremely rare condition characterized by idiopathic and progressive osteolysis affecting one or more bones [64,65][1][2]. The defining characteristic of GSD is the presence of both cortical and trabecular bone loss caused by progressive invasion of lymphatics into bone (Figure 1) [66][3]. While other CLAs only develop trabecular bone loss, GSD patients present both cortical and trabecular osteolysis. GSD typically involves one or few bones in a regional distribution. Functional consequences of GSD are determined by the anatomical location and progression of the disease. Major symptoms of GSD patients include bone pain, inflammation, limited movement, skeletal deformity, and chylothorax [67,68][4][5]. Chylothorax (accumulation of lymphatic fluid in the thoracic cavity) is a potentially life-threatening complication shared by all CLAs. Skull involvement and fractures are more prevalent in GSD compared with other CLAs [67,69][4][6]. Affected bone structures are filled with infiltrative soft tissue significantly more in GSD than other CLAs [66,68][3][5]. Chylothorax and spinal instability caused by progressive osteolysis is linked to 33–55% of GSD patient deaths [70][7]. The mortality of this condition has been shown to be around 13.3%, which indicates a relatively good prognosis due to spontaneous arrest of disease progression [71][8]. However, the prognosis becomes worse if the spine or thorax is involved due to the risk of neurological complications or chylothorax [71,72][8][9].
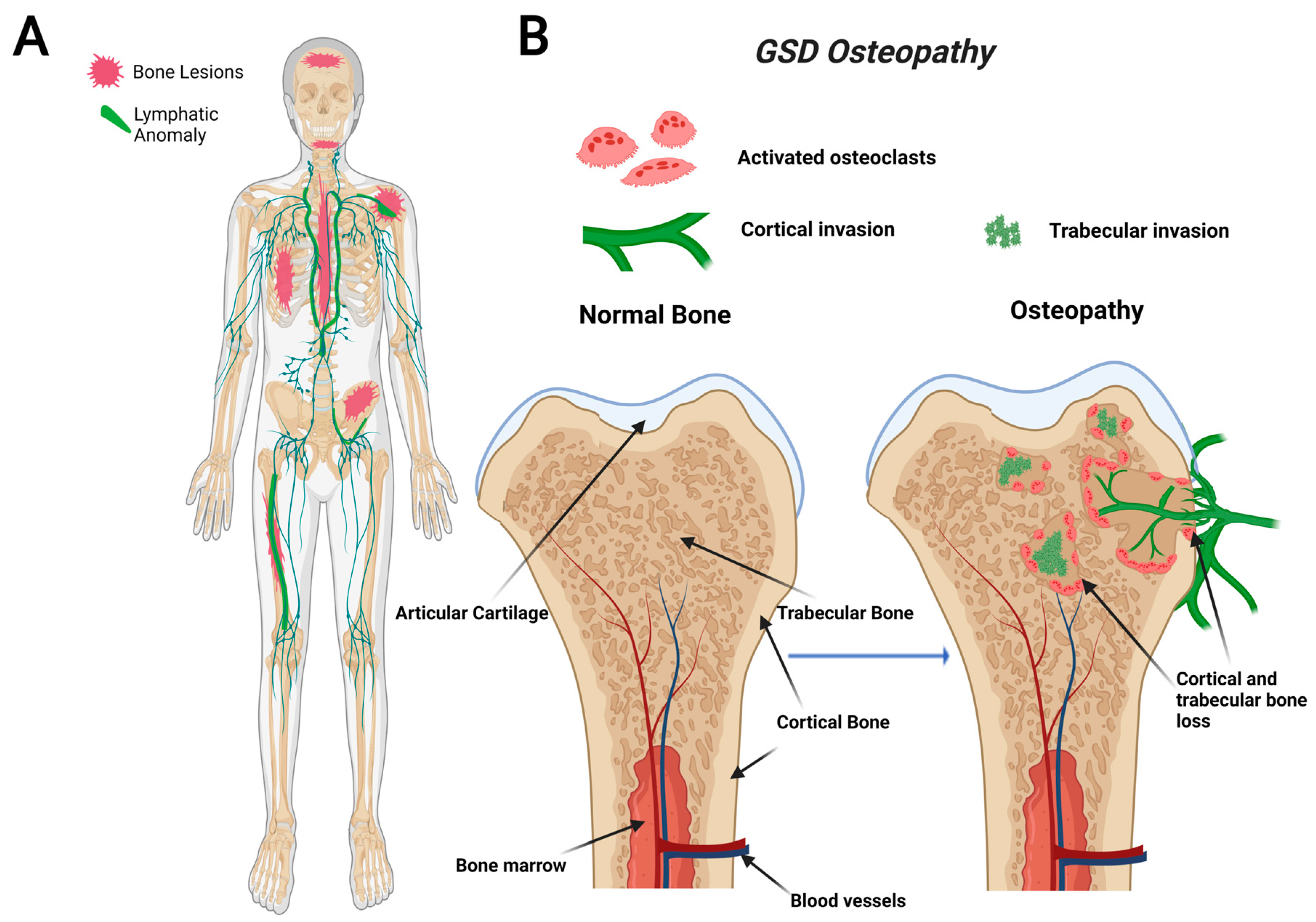
Figure 1. Visual representation of symptoms associated with Gorham-Stout Disease (GSD). GSD patients have regionally aggressive bone loss caused by abnormal lymphatic invasion (A). During GSD, both cortical and trabecular bone are resorbed due to lymphatic invasion (B).
Recently, a gain-of-function somatic mutation in KRAS (p.G12V) of GSD LECs has been shown to cause lymphatic malformations through hyperactivation of the RAS-MEK pathway [53,59,73][10][11][12]. This pathway is commonly implicated in abnormal lymphangiogenesis and several cancer types, including breast, colorectal, and liver [74][13]. Most recently, a mouse model with a similar activating somatic mutation in KRAS (p.G12D) was found to mimic the abnormal lymphatics and bone invasion identified in GSD patients [53,59][10][11]. Despite the recent advancements, further work is necessary to elucidate how this interaction between lymphatics and bone leads to progressive lymphatic growth and osteolysis.
2. General Lymphatic Anomaly (GLA)
GLA is characterized by diffuse lymphatic invasion with dilated lymphatic vessels that affect a variety of soft tissues and bone [54,66][3][14]. In contrast to GSD, GLA has a generalized distribution with multiorgan involvement containing lytic trabecular boney lesions (Figure 2). The axial skeleton is commonly affected in GLA with a focus on the ribs and spine. This variable presentation affecting multiple sites in the body makes it hard to diagnose due to the variety of symptoms varying from case to case. GLA can occur at any age, often presenting with the highest in children and teenagers with symptoms presenting before the age of 20 [68,75][5][15]. Thoracic involvement is a common and serious complication in patients diagnosed with GLA. A poorer prognosis is observed when there is a pleural effusion into soft tissues such as the lungs, heart, or peritoneum [68,76,77][5][16][17].
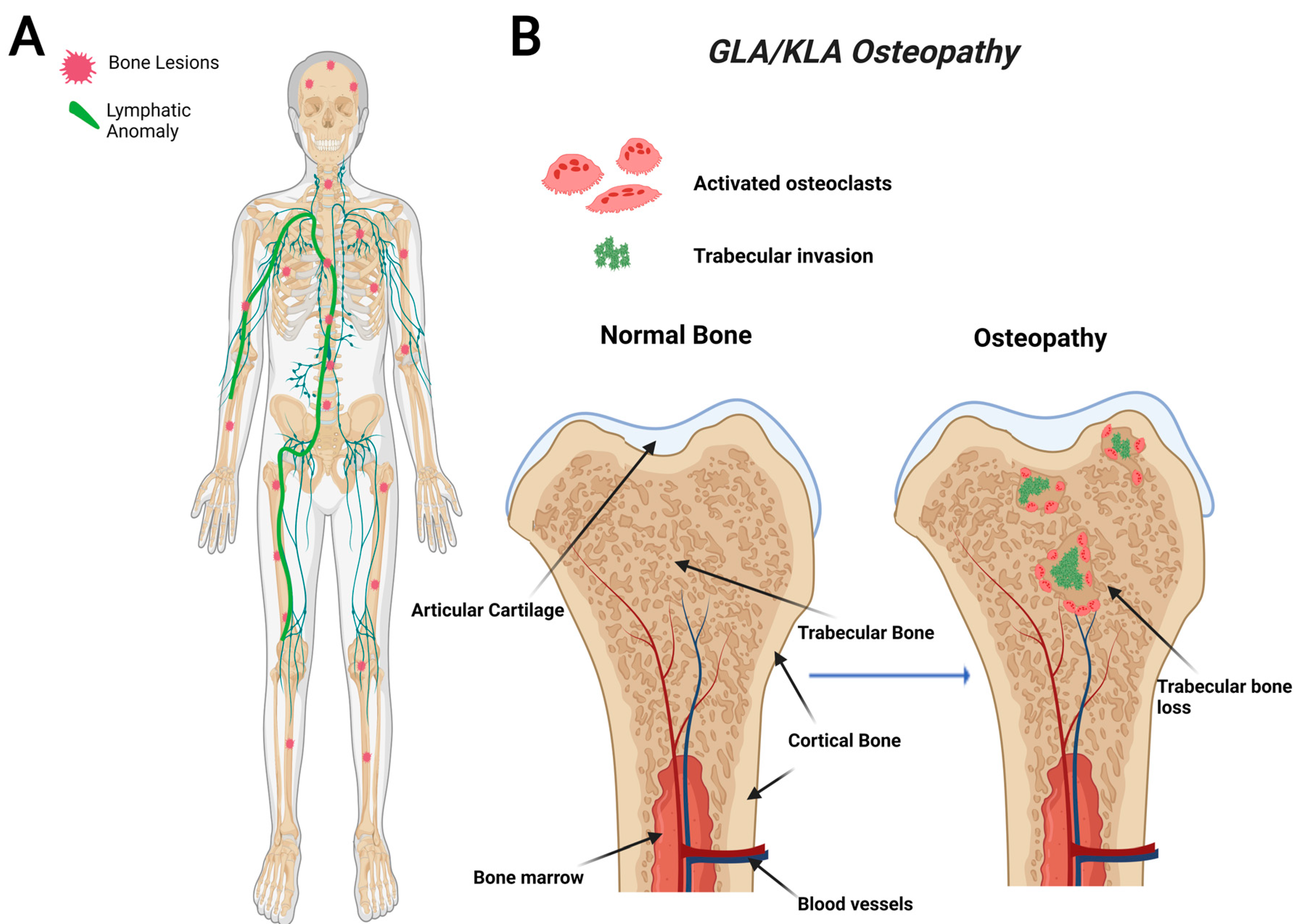
Figure 2. Visual representation of symptoms associated with Generalized Lymphatic Anomaly (GLA) and Kaposiform Lymphangiomatosis (KLA). GLA and KLA patients have generalized trabecular lytic lesions and lymphatic invasion in the thoracic cavity (A). Multifocal trabecular invasion is a common phenotype in GLA/KLA patients (B).
3. Kaposiform Lymphangiomatosis (KLA)
KLA is a particularly aggressive subtype of GLA that uniquely presents with spindle endothelial cells throughout the malformed lymphatic channels [66,78][3][18]. KLA displays a progressive involvement with severe morbidity and a high mortality rate. In a retrospective study of 20 patients between 1995 and 2011, the 5-year survival rate was reported to be 51%, with an overall survival rate of 34%; the cause of death in most instances is due to cardiorespiratory failure caused by consumptive coagulopathy [78][18]. This highly aggressive subtype presents at a young age, and many complications can arise, including organ failure, pleural and pericardial effusions, ascites, pain, and boney osteolysis [77][17].
The bones affected in KLA reside mainly in the thoracic cavity, and the bone involvement is similar to GLA (Figure 2). While KLA and GLA patients present with similar boney phenotypes, a defining feature that KLA displays its ability to affect multiple organs with pleural effusions, ultimately leading to respiratory distress in the thoracic cavity [68][5]. KLA diagnosis exhibits lymphatic channels with focal areas of spindle-shaped “kaposiform” endothelial cells, positive for Podoplanin and PROX-1 (markers for lymphatic tissue) [77][17]. KLA exhibits many features of GLA, such as nonprogressive lesions in bone’s medullary cavity, widespread lymphangiomatosis, and multifocal lymphatic malformations that involve the bones, viscera, thoracic, and abdominal cavities [42,78][18][19].
KLA is caused by mutations in the RAS viral oncogene homolog (NRAS) and Cbl Proto-oncogene (CBL) genes identified in patient tissue biopsies [49,57,60,77,79][17][20][21][22][23]. Because tissue biopsy may involve surgical intervention near vital organs that could lead to significant complications worsening the disease, the use of noninvasive diagnostics for KLA is warranted. Elevated Angiopoietin-2 (Ang-2) (a protein involved in blood vessel maturation and stability) was found to serve as a reliable biomarker for KLA [80,81][24][25]. In addition, Ang-2 levels normalize upon successful treatment suggesting its potential to track disease progression [82][26]. Interestingly, Ang-2 has been reported to have a negative effect on vessel maturation and stability [83][27]. Despite these discoveries, further research is necessary to determine Ang-2′s involvement in KLA pathogenesis.
4. Central Conducting Lymphatic Anomaly (CCLA)
CCLA, also known as channel-type LM, is a rare subset of disorders that are caused by dysfunction of the thoracic duct and/or cisternae chylae. These result in reflux and leakage of lymphatic fluid around the lungs, heart, abdomen, and legs, often with lymphedema involving bilateral lower extremities [84][28]. CCLA’s distinguishing clinical feature consists of dilated central conducting lymphatic channels that lead to improper fluid transport (Figure 3). Common complications include chylothorax, pulmonary lymphangiectasia, chylous ascites, protein-losing enteropathy, lymphedema, cutaneous vesicles, or superficial chylous leaks [85][29]. Boney lesions have also been reported in patient vertebrae [84][28]. These osseous changes include focal areas of hyperlucency due to dilated intraosseous lymphatic channels with a more permeative appearance (unpublished observations and personal communication with Dr. Michael Kelly). CCLA can occur at any age; however, the majority of cases are reported in pediatric patients below the age of 20. CCLA has been recently classified as a CLA due to multiorgan invasive profile, yet it is commonly referred to as channel-type lymphatic malformation.
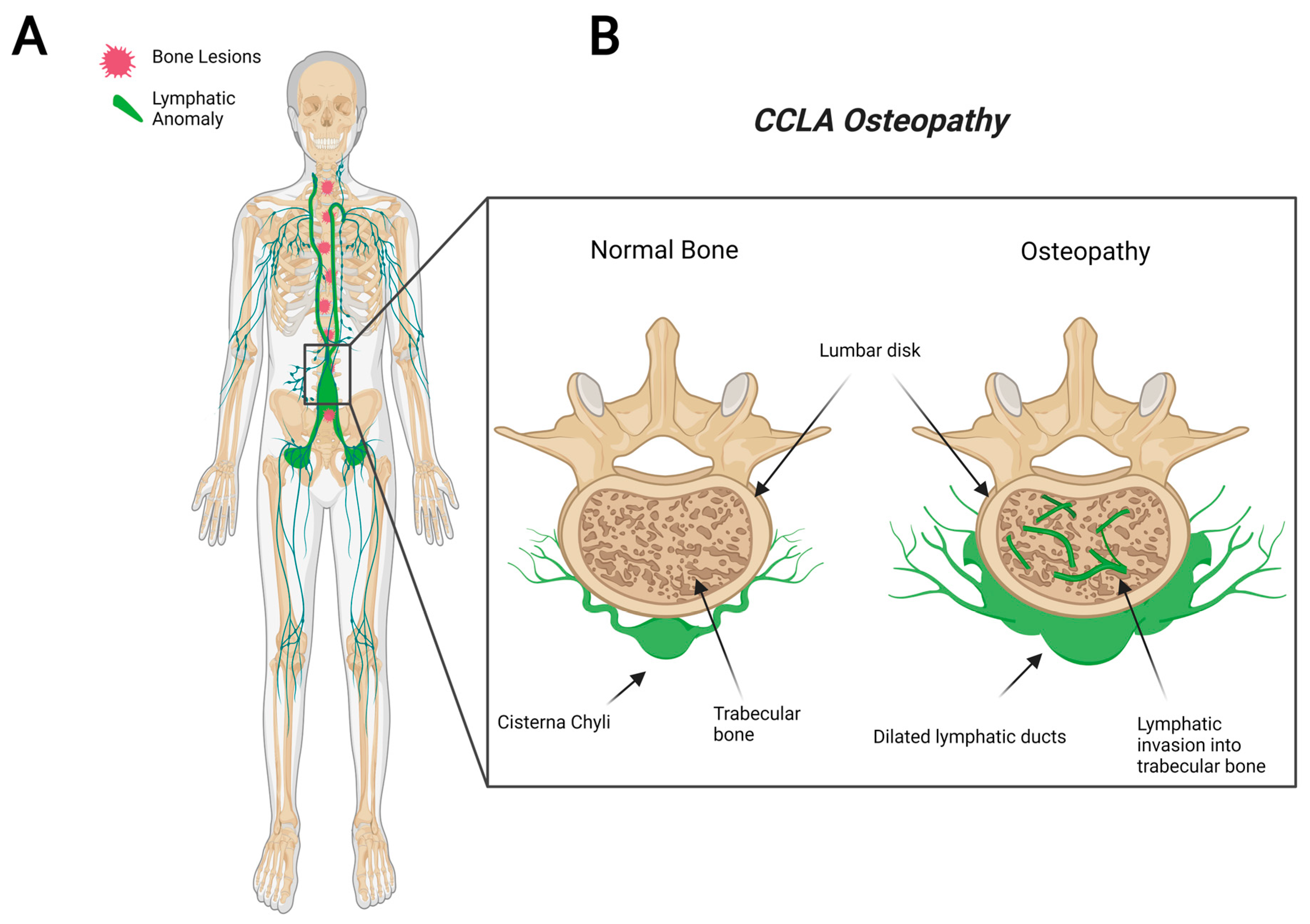
Figure 3. Visual representation of symptoms associated with Central Conducting Lymphatic Anomaly. CCLA patients fail to transport lymphatic fluid and suffer from dilated thoracic ducts. Dilated ducts invade proximal tissue and can cause boney lesions (A). Channel-like osseous lesions are found in the trabecular bones (B).
References
- Liu, Y.; Zhong, D.-R.; Zhou, P.-R.; Lv, F.; Ma, D.-D.; Xia, W.-B.; Jiang, Y.; Wang, O.; Xing, X.-P.; Li, M. Gorham-Stout disease: Radiological, histological, and clinical features of 12 cases and review of literature. Clin. Rheumatol. 2014, 35, 813–823.
- Gorham, L.W.; Stout, A.P. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): Its relation to hemangiomatosis. J. Bone Jt. Surg. Am. Vol. 1955, 37-a, 985–1004.
- Kato, H.; Ozeki, M.; Fukao, T.; Matsuo, M. MR imaging findings of vertebral involvement in Gorham-Stout disease, generalized lymphatic anomaly, and kaposiform lymphangiomatosis. Jpn. J. Radiol. 2017, 35, 606–612.
- Rossi, M.; Buonuomo, P.S.; Battafarano, G.; Conforti, A.; Mariani, E.; Algeri, M.; Del Fattore, A.; D’Agostini, M.; Macchiaiolo, M.; De Vito, R.; et al. Dissecting the mechanisms of bone loss in Gorham-Stout disease. Bone 2020, 130, 115068.
- Ozeki, M.; Fujino, A.; Matsuoka, K.; Nosaka, S.; Kuroda, T.; Fukao, T. Clinical Features and Prognosis of Generalized Lymphatic Anomaly, Kaposiform Lymphangiomatosis, and Gorham-Stout Disease. Pediatr Blood Cancer 2016, 63, 832–838.
- Dellinger, M.T.; Garg, N.; Olsen, B.R. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone 2014, 63, 47–52.
- Tena-Sanabria, M.E.; Jesús-Mejenes, L.Y.; Fuentes-Herrera, G.; Álvarez-Martínez, F.A.; Victorio-García, N.P.; Núñez-Enríquez, J.C. A report of two children with Gorham-Stout disease. BMC Pediatr. 2019, 19, 206.
- Florchinger, A.; Bottger, E.; Claass-Bottger, F.; Georgi, M.; Harms, J. Gorham-Stout syndrome of the spine. Case report and review of the literature. Rofo 1998, 168, 68–76.
- Perschbacher, S.E.; Perschbacher, K.A.; Pharoah, M.J.; Bradley, G.; Lee, L.; Yu, E. Gorham’s disease of the maxilla: A case report. Dentomaxillofac. Radiol. 2010, 39, 119–123.
- Sepehr, N.H.; McCarter, A.L.; Helaers, R.; Galant, C.; Boon, L.M.; Brouillard, P.; Vikkula, M.; Dellinger, M.T. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight 2021, 6, e149831.
- Nozawa, A.; Ozeki, M.; Niihori, T.; Suzui, N.; Miyazaki, T.; Aoki, Y. A somatic activating KRAS variant identified in an affected lesion of a patient with Gorham–Stout disease. J. Hum. Genet. 2020, 65, 995–1001.
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293.
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198.
- Manevitz-Mendelson, E.; Leichner, G.S.; Barel, O.; Davidi-Avrahami, I.; Ziv-Strasser, L.; Eyal, E.; Pessach, I.; Rimon, U.; Barzilai, A.; Hirshberg, A.; et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis 2018, 21, 287–298.
- Alvarez, O.A.; Kjellin, I.; Zuppan, C.W. Thoracic Lymphangiomatosis in a Child. J. Pediatric Hematol./Oncol. 2004, 26, 136–141.
- Yabuta, M.; Nozaki, T.; Fukuda, T.; Suzuki, K.; Kurihara, Y.; Niimi, Y. Generalized Lymphatic Anomaly Associated with Multiple Paraspinal Arteriovenous Malformations and Renal Artery Aneurysms. J. Vasc. Interv. Radiol. 2018, 29, 1633–1635.e1.
- Ricci, K.W.; Iacobas, I. How we approach the diagnosis and management of complex lymphatic anomalies. Pediatr. Blood Cancer 2021, 2021, e28985.
- Croteau, S.E.; Kozakewich, H.P.; Perez-Atayde, A.R.; Fishman, S.J.; Alomari, A.I.; Chaudry, G.; Mulliken, J.B.; Trenor, C.C., 3rd. Kaposiform Lymphangiomatosis: A Distinct Aggressive Lymphatic Anomaly. J. Pediatr. 2013, 164, 383–388.
- Iacobas, I.; Adams, D.M.; Pimpalwar, S.; Phung, T.; Blei, F.; Burrows, P.; Lopez-Gutierrez, J.C.; Levine, M.A.; Trenor, C.C., III. Multidisciplinary guidelines for initial evaluation of complicated lymphatic anomalies—Expert opinion consensus. Pediatric Blood Cancer 2020, 67, e28036.
- Rodriguez-Laguna, L.; Agra, N.; Ibáñez, K.; Oliva-Molina, G.; Gordo, G.; Khurana, N.; Hominick, D.; Beato, M.; Colmenero, I.; Herranz, G.; et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J. Exp. Med. 2018, 216, 407–418.
- Martinez-Corral, I.; Zhang, Y.; Petkova, M.; Ortsäter, H.; Sjöberg, S.; Castillo, S.D.; Brouillard, P.; Libbrecht, L.; Saur, D.; Graupera, M.; et al. Blockade of VEGF-C signaling inhibits lymphatic malformations driven by oncogenic PIK3CA mutation. Nat. Commun. 2020, 11, 2869.
- Blesinger, H.; Kaulfuß, S.; Aung, T.; Schwoch, S.; Prantl, L.; Rössler, J.; Wilting, J.; Becker, J. PIK3CA mutations are specifically localized to lymphatic endothelial cells of lymphatic malformations. PLoS ONE 2018, 13, e0200343.
- Le Cras, T.D.; Goines, J.; Lakes, N.; Pastura, P.; Hammill, A.M.; Adams, D.M.; Boscolo, E. Constitutively active PIK3CA mutations are expressed by lymphatic and vascular endothelial cells in capillary lymphatic venous malformation. Angiogenesis 2020, 23, 425–442.
- Le Cras, T.D.; Mobberley-Schuman, P.S.; Broering, M.; Fei, L.; Trenor, C.C., 3rd; Adams, D.M. Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis 2017, 20, 163–173.
- Ozeki, M.; Nozawa, A.; Kawamoto, N.; Fujino, A.; Hirakawa, S.; Fukao, T. Potential biomarkers of kaposiform lymphangiomatosis. Pediatr. Blood Cancer 2019, 66, e27878.
- Crane, J.; Manfredo, J.; Boscolo, E.; Coyan, M.; Takemoto, C.; Itkin, M.; Adams, D.M.; Le Cras, T.D. Kaposiform lymphangiomatosis treated with multimodal therapy improves coagulopathy and reduces blood angiopoietin-2 levels. Pediatr. Blood Cancer 2020, 67, e28529.
- Zheng, W.; Nurmi, H.; Appak, S.; Sabine, A.; Bovay, E.; Korhonen, E.A.; Orsenigo, F.; Lohela, M.; D’Amico, G.; Holopainen, T.; et al. Angiopoietin 2 regulates the transformation and integrity of lymphatic endothelial cell junctions. Genes Dev. 2014, 28, 1592–1603.
- Ricci, K.W.; Hammill, A.M.; Mobberley-Schuman, P.; Nelson, S.C.; Blatt, J.; Bender, J.L.G.; McCuaig, C.C.; Synakiewicz, A.; Frieden, I.J.; Adams, D.M. Efficacy of systemic sirolimus in the treatment of generalized lymphatic anomaly and Gorham–Stout disease. Pediatr. Blood Cancer 2019, 66, e27614.
- Trenor, C.C.; Chaudry, G. Complex lymphatic anomalies. Semin. Pediatr. Surg. 2014, 23, 186–190.
More